Article Text

Abstract
Background Hypertrophic cardiomyopathy (HCM) is frequently fatal in infancy. Mitochondrial disease causing infantile HCM is characterised by extreme biochemical and genetic heterogeneity, but deficiency of respiratory chain complex I is observed relatively frequently. Identification of the precise genetic basis has prognostic implications for the likelihood of neurological involvement.
Objective The authors' objective is to report two heterozygous missense mutations in the NDUFAF1 gene as a cause of fatal infantile HCM in a patient with isolated complex I deficiency.
Methods The authors investigated a cohort of 30 paediatric patients with complex I deficiency using biochemical and genetic approaches. The patients were clinically heterogeneous; phenotypes included HCM, Leigh syndrome, other encephalomyopathies and multisystem disease. Complex I assembly was evaluated using Blue Native polyacrylamide gel electrophoresis.
Results Sequence analysis of NDUFAF1 revealed compound heterozygous missense mutations (c.631C>T;p.Arg211Cys and c.733G>A;p.Gly245Arg) in one patient with fatal infantile HCM. These changes were absent in 240 ethnically matched control alleles. No NDUFAF1 mutations were observed in the remaining patients. Functional studies demonstrated a severe reduction in NDUFAF1 protein in Western blots of patient fibroblasts and accumulation of abnormal complex I assembly intermediates on Blue Native polyacrylamide gel electrophoresis.
Conclusions The authors report a case of fatal infantile HCM caused by missense mutations in NDUFAF1 associated with complex I misassembly. Establishing a genetic diagnosis in mitochondrial cardiomyopathy is challenging and achieved in only a minority of cases because of complex genetics. A precise genetic diagnosis is important to provide accurate prognostic and genetic counselling advice regarding recurrence risks and to guide future reproductive options.
- Mitochondrial respiratory chain
- hypertrophic cardiomyopathy
- NADH:ubiquinone oxidoreductase
- complex I deficiency
- NDUFAF1
- molecular genetics
- nutrition and metabolism
- neuromuscular disease
- muscle disease
- molecular genetics
Statistics from Altmetric.com
- Mitochondrial respiratory chain
- hypertrophic cardiomyopathy
- NADH:ubiquinone oxidoreductase
- complex I deficiency
- NDUFAF1
- molecular genetics
- nutrition and metabolism
- neuromuscular disease
- muscle disease
- molecular genetics
Introduction
Cardiomyopathy is a rare but potentially devastating disorder of the heart muscle with an incidence of 1.13 cases per 100 000 children per year in the USA.1 Hypertrophic cardiomyopathy (HCM) accounts for ∼6% of cases listed for paediatric heart transplantation.2 More than two-thirds of children do not have a known diagnosis at the time of clinical presentation with cardiomyopathy. The differential diagnosis includes viral illness, genetic defects of sarcomeric proteins3 and inherited metabolic diseases.4 Approximately 9% of paediatric HCM cases are caused by an inborn error of metabolism,5 including lysosomal storage disorders, fat oxidation defects and mitochondrial respiratory chain enzyme deficiencies (the largest subgroup, representing over half of metabolic HCM cases).4 Diagnosis rests on clinical characterisation, family history, echocardiographic appearances and laboratory investigations, including tissue biopsy.
Identification of the precise genetic basis of HCM yields essential prognostic information. The likelihood of progressive neurological disease is critically important, since many centres would consider this a contraindication to cardiac transplantation. Disorders of the mitochondrion are characterised by extreme genetic heterogeneity, with multiple possible modes of inheritance related to the bigenomic (mitochondrial and nuclear) inheritance of this dynamic subcellular organelle.6 Mitochondrial disease can theoretically arise from the mutation of any of the >1500 components of the mitochondrial proteome,7 although, so far, only ∼80 nuclear genes have been proven to cause mitochondrial dysfunction,8 in addition to the 37 mitochondrially encoded genes.
Mitochondrial complex I (NADH:ubiquinone oxidoreductase; EC 1.6.5.3) is the largest enzyme in the respiratory chain, with essential functions in electron transfer and proton pumping. Complex I generates ∼40% of the proton motive force that is eventually harnessed by ATP synthase (complex V) to synthesise ATP from ADP and inorganic phosphate.9 10 Human complex I consists of 38 different nuclear-encoded structural proteins, 7 mitochondrial DNA (mtDNA)-encoded structural proteins, 1 flavin mononucleotide moiety and 8 iron–sulphur clusters, all of which are assembled together in an intricate process that remains incompletely understood.11 Complex I deficiency is the most commonly observed biochemical defect in childhood-onset mitochondrial disease and is genetically heterogeneous. Fewer than half of cases appear to be caused by mutations in structural subunits of the enzyme, and mutations in assembly factors are thought to account for the majority of cases.12 Mutations in nine different assembly factors (NDUFAF1,13 NDUFAF2,14 NDUFAF3,15 NDUFAF4,16 C8orf38,7 C20orf7,17 FOXRED1,12 NUBPL18 and ACAD919) have been associated with human complex I deficiency to date.
Here, we used a combined biochemical and genetic approach to identify compound heterozygous missense mutations in NDUFAF1 as a novel cause of complex-I-deficient fatal infantile HCM. NDUFAF1 encodes a protein previously known as CIA30 that is essential for the correct assembly of complex I, although its precise function remains unclear.11 20 21
Patients and methods
Clinical data
The patient, the first child of healthy unrelated French parents, was born at 37 weeks of gestation weighing 3.17 kg. The neonatal period was unremarkable, and early developmental milestones were normal. She was first presented to medical attention at 6 months with an episode of respiratory-syncytial-virus-positive bronchiolitis, which was managed conservatively with nasogastric tube feeding. Following discharge, she became more unwell, with pallor, poor feeding, sweating and weight loss of 380 g. At 6.5 months, she presented to the emergency department in cardiogenic shock (O2 saturation of 90%, heart rate of 180 beats/min and hepatomegaly palpable at 5 cm below the inferior costal margin). Initial blood gas values revealed metabolic acidosis (pH 6.86; base deficit, 22 mmol/l). Before retrieval to the regional cardiac intensive care unit, she was intubated and ventilated; given intraosseous volume replacement, antibiotics and bicarbonate; and started on dopamine infusion.
Echocardiogram (figure 1A,B) demonstrated pericardial effusion, biventricular hypertrophy and mild to moderate left ventricular (LV) dysfunction (fractional shortening, 25%). Both atria were enlarged, and there was evidence of diastolic impairment. Apical views showed trabeculation suggestive of non-compaction of the left ventricle (NCLV). Coronary artery anatomy was normal. Despite positive pressure ventilation and inotropic support with dopamine and adrenaline, she remained hypotensive and acidotic (pH 7.2; base deficit, 19 mmol/l), with an elevated blood lactate level (9 mmol/l; reference, <2 mmol/l). The acidosis did not improve, and cardiac function continued to deteriorate. Veno-arterial extracorporeal membrane oxygenation (ECMO) was commenced on day 2 of her critical illness. Subsequently, balloon atrial septostomy was performed to decompress the left atrium, and pericardiocentesis was undertaken. Lactic acidosis persisted on full ECMO support (flow, 165%), despite the patient being warm and well perfused. Haemofiltration was therefore commenced via the ECMO circuit on the following day, with a reduction in blood lactate levels to 2–7 mmol/l. ECMO support was successfully weaned and stopped after 9 days. Continuous veno-venous haemofiltration was continued further for 6 days.

Short-axis echocardiography showing ventricular hypertrophy with trabeculations (indicated by the arrows) suggestive of NCLV and posterior pericardial effusion (A). Long-axis echocardiography showing ventricular hypertrophy, thickened LV papillary muscles (indicated by the asterisk), unusual speckled appearance of the myocardium and bright subendocardium, particularly in the septum of the left ventricle (septum indicated by the double arrow) (B). Ultrastructural examination of muscle demonstrates accumulation of enlarged and abnormal mitochondria, which show electron-dense cristae forming concentric arrays (C). LV myocardium showing myocardial hypertrophy without evidence of disarray but with cytoplasmic clearing and occasional small round eosinophilic inclusions, which represent giant mitochondria (D,E). Photomicrographs demonstrating a liver with marked zonal perivenular macrovesicular steatosis (F,G). NCLV, non-compaction of the left ventricle; LV, left ventricular.
The combination of echocardiographic appearances and persistent lactic acidosis on full ECMO support led to the suspicion of an inborn error of metabolism. Urine organic acid analysis demonstrated marked excretion of lactate, pyruvate, ketones and Krebs cycle intermediates (fumarate, malate and 2-oxoglutarate), as well as moderately increased levels of glutarate and 3-hydroxyglutarate. This profile was consistent with, although not diagnostic of, a mitochondrial respiratory chain disorder. Plasma acylcarnitine analysis revealed moderately elevated hydroxybutyrylcarnitine but no other abnormalities. Cranial ultrasound results were normal, and electroencephalographic findings were consistent with a sedated critically unwell infant.
Muscle biopsy showed no ragged-red or cytochrome-c-oxidase-negative fibres, but there was increased lipid deposition. Electron microscopy demonstrated accumulation of enlarged and abnormal mitochondria (figure 1C). Spectrophotometric analysis of respiratory chain enzyme activities revealed severe isolated deficiency of complex I in skeletal muscle, with 25% residual activity compared to the lowest control (patient complex I activity, 0.026; reference range, 0.104–0.268 (ratio to citrate synthase activity)). The activities of complexes II+III and IV were normal. Forty-eight hours after continuous veno-venous haemofiltration was stopped, blood lactate levels rose to 17–18 mmol/l despite maximal medical treatment for low cardiac output, including inotropes, cooling, sedation and muscle relaxation. Cardiac reserve was minimal. On day 24 of her critical illness, she had cardiorespiratory arrest requiring two doses of epinephrine. Output was briefly re-established, but she died later that night.
Postmortem examination revealed an enlarged globular heart (weight, 93 g; weight expected for age, 34 g). Sections from the myocardium showed myocardial hypertrophy without evidence of disarray, but with foci of myofibre loss and replacement fibrosis, focal haemorrhage and haemosiderin deposition within the centres of the LV papillary muscles, and a mild degree of LV endocardial fibrosis (figure 1D,E). There was no myocarditis. Liver histology revealed marked zonal macrovesicular steatosis (figure 1F,G). Respiratory chain enzyme activities in the liver were normal (complex I, 0.207; reference range, 0.054–0.221).
Patient cohort
We investigated a cohort of 30 paediatric patients with complex I deficiency using biochemical and genetic approaches. These patients were clinically heterogeneous: phenotypes included Leigh syndrome, other encephalomyopathies, HCM, isolated myopathy and multisystem disease. mtDNA analysis revealed pathogenic mutations in 10 cases (33%). Complex I assembly in the remaining cases was investigated using Blue Native polyacrylamide gel electrophoresis (BN-PAGE). A candidate gene approach was used to search for assembly factor mutations in patients accumulating abnormal complex I subassemblies on BN-PAGE analysis. NDUFS4 was also screened since mutations in this subunit are known to disrupt complex I assembly.
Genetic analyses
All samples were taken after informed patient/parental consent had been obtained, and the study was approved by the local ethics committee. Total DNA was extracted from the muscle of the patient and from the blood of the parents and control subjects. The entire mtDNA, exons and flanking intronic regions of the nuclear genes NDUFS4, FOXRED1 and NDUFAF1 (GenBank accession numbers NM_002495, NM_017547 and NM_016013.2, respectively) were amplified using specific primers (sequences available on request). Direct sequencing was performed using the BigDye® Terminator v1.1 Cycle Sequencing Kit (Applied Biosystems). Total RNA was extracted from cultured skin fibroblasts from the patient using the RNeasy Tissue Kit (Qiagen). Complementary DNA was synthesised, and the full-length transcript was amplified and sequenced.
In vitro studies of mutant NDUFAF1
Complex I activity of fibroblasts was measured with a microplate assay kit according to the manufacturer's instructions, and the results were normalised to complex IV activity assayed using the same microplate method (MitoSciences, Eugene, Oregon, USA). All measurements were performed in triplicate.
Expression of NDUFAF1 was assessed in mitoplasts derived from patient and control fibroblasts and 143B206 ρ0 cells lacking mitochondrially encoded proteins22 with Western blot analysis, as previously described,23 using a monoclonal anti-NDUFAF1 antibody (Novus Biologicals, Cambridge, UK). A monoclonal anti-SDHA antibody (MitoSciences) was used as a loading control. Levels of mtDNA and nuclear-encoded subunits of complex I were investigated using antibodies to ND1 (a kind gift from Dr A Lombès) and NDUFA9 (MitoSciences), respectively.
The effect of the NDUFAF1 mutations on complex I holoenzyme assembly was investigated in patient and control mitoplasts isolated from fibroblasts, performed as previously described, using BN-PAGE.12 24 Complex I assembly was assessed using antibodies to the ND1, NDUFB6 and NDUFA9 subunits; the other mitochondrial enzyme complexes were probed with antibodies to SDHA (complex II), UQCR2 (complex III) and ATP5A1 (complex V) (MitoSciences). No cardiac or skeletal muscle samples remained for use in functional studies.
Results
Identification of mutations in NDUFAF1
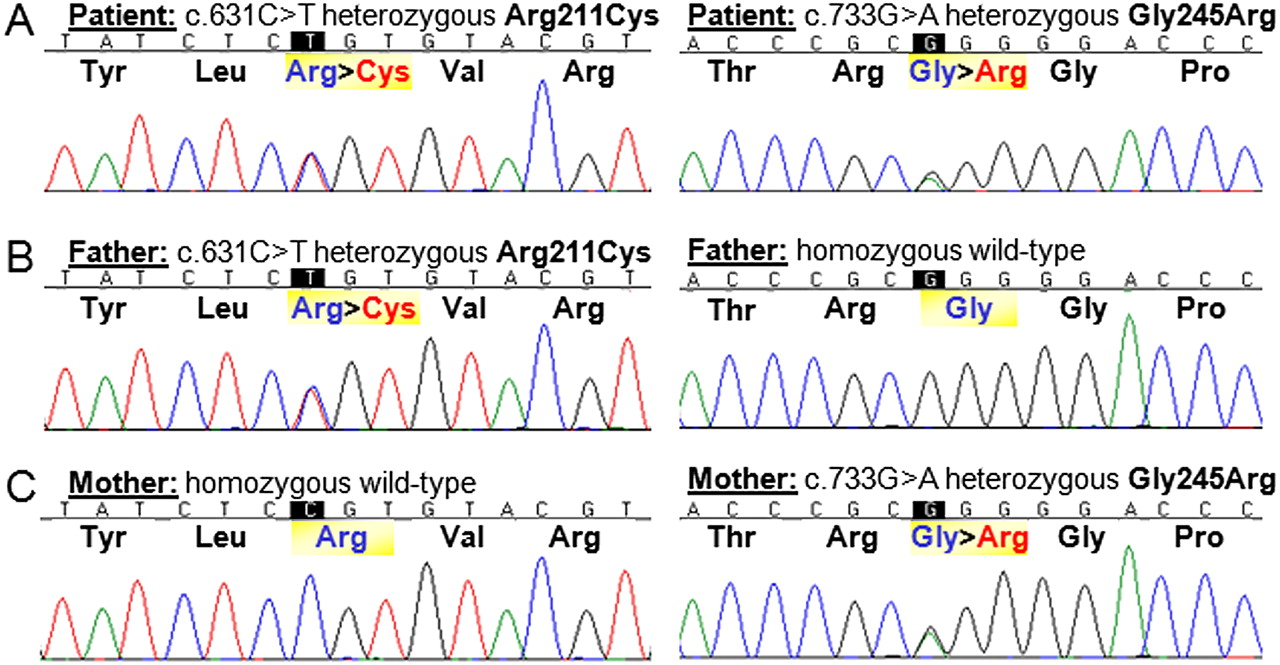
Sequence analysis of NDUFAF1 revealed two heterozygous missense mutations in exon 2 in the patient: c.631C>T;p.Arg211Cys and c.733G>A;p.Gly245Arg (figure 2A), which were also confirmed in complementary DNA. Each parent was heterozygous for one of these mutations (figure 2B,C), confirming that they were on separate alleles (ie, in trans) in the patient. Phylogenetic analysis demonstrated that Arg211 and Gly245 are highly conserved amino acids (figure 3). These changes were absent in 240 ethnically matched control alleles and also in 1094 control individuals genotyped in the 1000 Genomes cohort (http://www.1000genomes.org). No NDUFAF1 mutations were observed in the remainder of the patient cohort. In the patient, no mutations in the mtDNA or in NDUFS4 or FOXRED1 (two nuclear genes previously associated with complex-I-deficient cardiomyopathy) were observed.
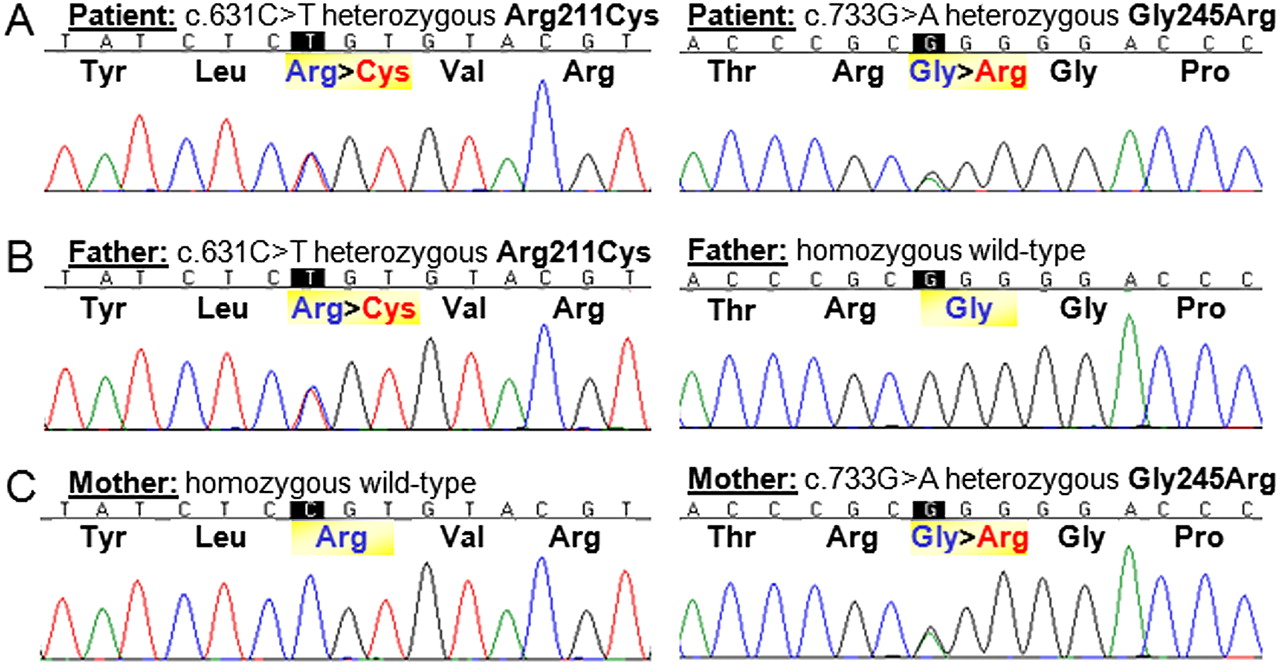
Sequencing of exon 2 of the NDUFAF1 gene revealed two compound heterozygous mutations, c.631C>T;Arg211Cys and c.733G>A;Gly245Arg, in the patient (A). Her father is heterozygous for the c.631C>T;Arg211Cys mutation and homozygous wild type for the second variant (B), while her mother is heterozygous for the c.733G>A;Gly245Arg mutation and homozygous wild type for the first mutation (C).

The amino acid residues affected by the missense mutations in exon 2 are 33 amino acids apart within the NDUFAF1 protein and are highly conserved from humans to the nematode Caenorhabditis elegans and fungi.
Functional characterisation of the NDUFAF1 mutations
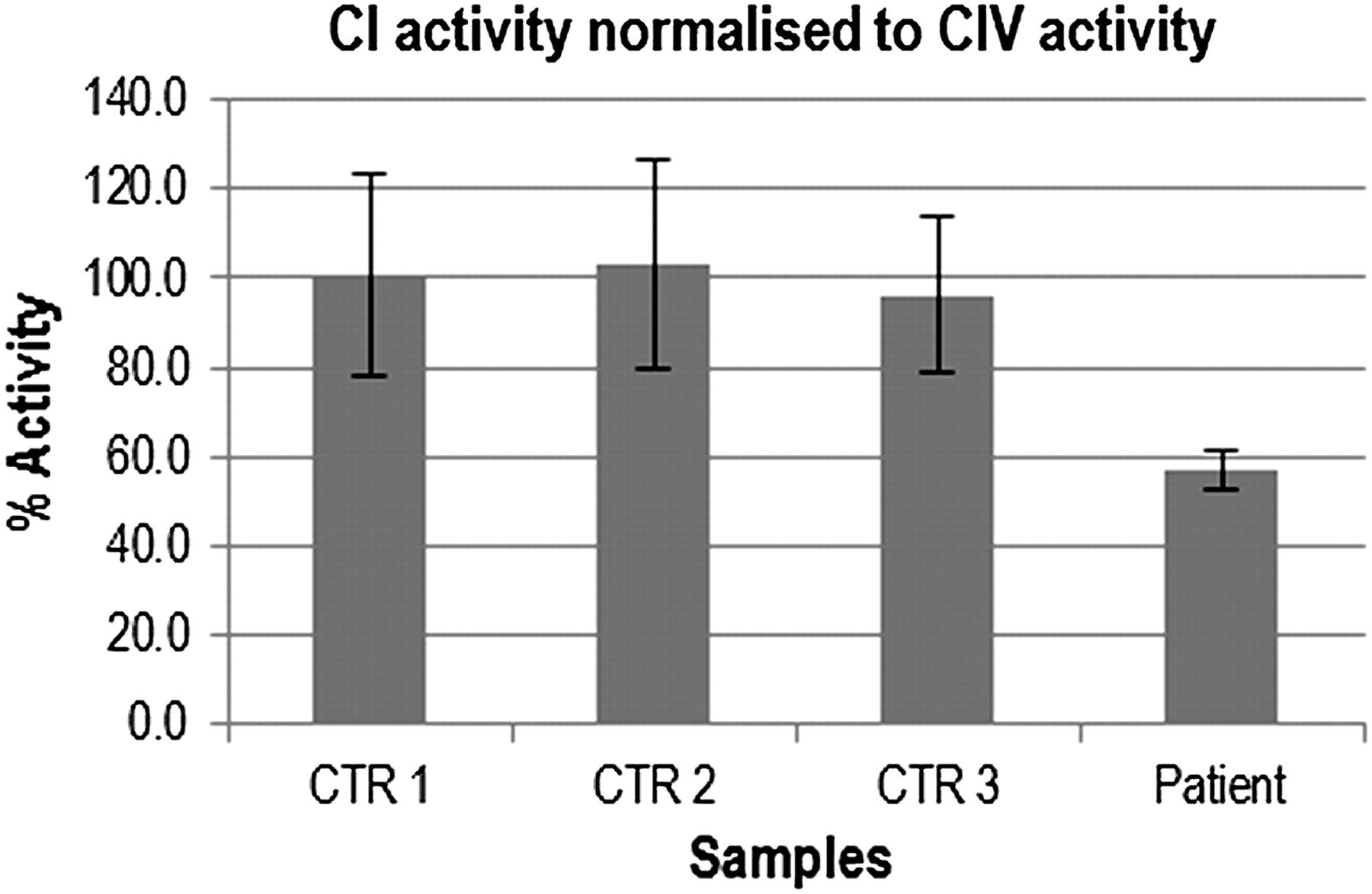
Patient fibroblasts showed ∼57% residual complex I activity compared to three age-matched normal controls (figure 4). NDUFAF1 protein levels were severely reduced in patient mitoplasts compared to three controls and ρ0 cells on Western blot analysis (figure 5A). Levels of the mitochondrially encoded complex I core subunit ND1 were normal in the patient, whereas levels of the nuclear-encoded membrane arm subunit NDUFA9 were reduced.

Complex I activity was measured in patient and CTR fibroblasts using the microplate assay kit. All measurements were performed in triplicate and expressed as ratio to complex IV activity assayed with the same method. The residual complex I activity in the patient was ∼57% of the CTR mean in cultured skin fibroblasts (compared to 25% of the lowest CTR in skeletal muscle, as measured by spectrophotometric assay; data not shown). CTR, control.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
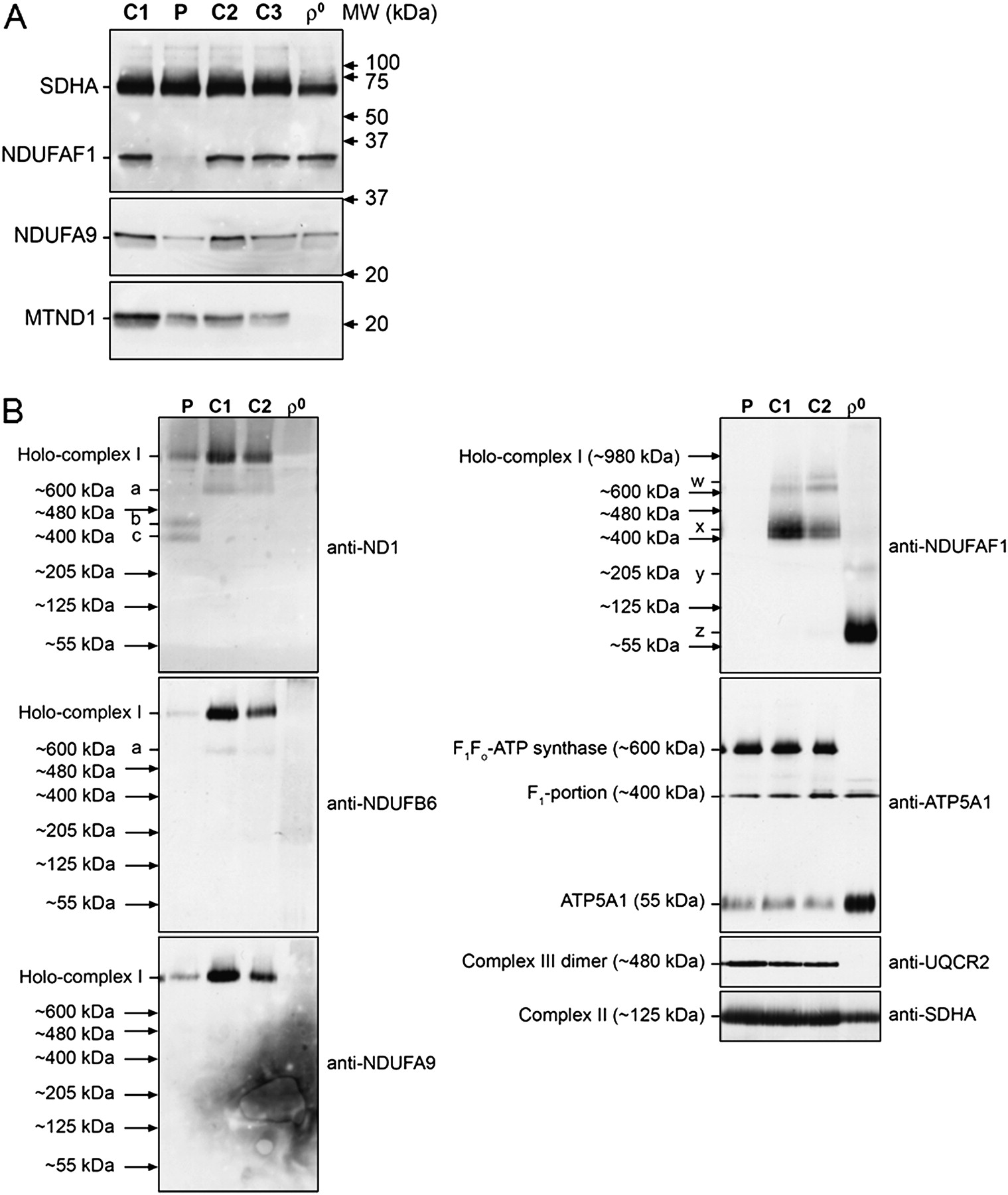
Western blot analysis demonstrated a severe reduction in NDUFAF1 protein levels in P mitoplasts compared to C mitoplasts isolated from fibroblasts; normal staining of SDHA confirmed equal loading of all samples. Levels of ND1 (a core subunit assembled early into the nascent complex I enzyme) were normal, whereas levels of NDUFA9 (a membrane arm subunit that was incorporated later) were reduced in patients. As expected, ρ0 cells lacked mitochondrially encoded ND1 but had normal levels of all nuclear-encoded proteins analysed (A). BN-PAGE of mitoplasts isolated from P and C fibroblasts and ρ0 cells. Complex I was detected using antibodies to the ND1, NDUFB6 and NDUFA9 subunits. Levels of the ∼980 kDa complex I holoenzyme were severely reduced in P mitoplasts. In contrast, levels of complexes II, III and V, detected with antibodies to SDHA, UQCR2 and ATP5A1, respectively, were the same in the P and C lanes, confirming equal loading of the samples. The signals of these antibodies were used as molecular weight markers. Abnormal complex I assembly intermediates of ∼400 kDa (c) and ∼460 kDa (b) were seen to accumulate in the patient—but a physiological subassembly at ∼600 kDa (a) present in controls was not seen in the patient—when probing against ND1 (top left). No NDUFAF1 signal could be detected in the P sample (top right), confirming the findings of Western blot analysis. Probing against NDUFAF1 revealed further physiological subassemblies in C cells: a doublet at ∼460 kDa (x) and two subassemblies at >600 kDa (w). These were not observed in P mitoplasts. No fully assembled complex I holoenzyme was observed in ρ0 cells, as expected; however, when probing against NDUFAF1, we saw a very small subassembly of ∼80 kDa (z) and a less visible band at ∼205 kDa (y) (B). BN-PAGE, Blue Native polyacrylamide gel electrophoresis. P, patient; C, control.
BN-PAGE analysis demonstrated a severe reduction in the ∼980 kDa complex I holoenzyme in the patient, with all complex I subunit antibodies used (figure 5B). In contrast, the levels of complex II, complex III and complex V holoenzymes appeared to be normal. Distinctive complex I subassemblies, corresponding to ∼400 and ∼460 kDa, were observed in the patient when probing against ND1 (figure 5B). These were not seen in any of the control samples. In contrast, a physiological subassembly of ∼600 kDa was only present in control samples (figure 5B). No signal was detected in the patient sample when probing against NDUFAF1, suggesting that the very low level of residual NDUFAF1 protein in the patient is not able to efficiently bind the nascent complex I, leaving it stalled at an early assembly stage.11 13 Further physiological subassemblies were observed in the controls when probing against NDUFAF1: a doublet at ∼460 kDa, corresponding to the NDUFAF1 entry point in the complex I assembly process,11 and two other larger subassemblies of >600 kDa. Interestingly, ρ0 cells did not contain any fully assembled complex I holoenzyme, as expected; however, when probing against NDUFAF1, we saw a very small subassembly of ∼80 kDa, together with a less visible band at ∼205 kDa, indicating that the complex I assembly process in these cells lacking mitochondrially encoded subunits, presumably containing only a few nuclear-encoded subunits, is disrupted at an extremely early stage.
Discussion
We employed a combined biochemical and genetic approach, using BN-PAGE to identify patients accumulating abnormal complex I assembly intermediates in cultured skin fibroblasts, followed by candidate gene sequencing of known complex I assembly factors to identify compound heterozygous missense mutations in NDUFAF1 as a novel cause of fatal infantile HCM. We observed NDUFAF1 mutations in one of our cohort of 30 paediatric patients with isolated complex I deficiency associated with heterogeneous clinical phenotypes. The affected patient was a 7-month-old infant who was previously well and developing normally prior to the onset of her cardiac symptoms. The pathogenicity of the NDUFAF1 mutations is supported by several lines of evidence. The healthy parents were each heterozygous for one of the missense mutations observed in their daughter, and these mutations were not present in 120 control subjects and 1094 individuals genotyped for the 1000 Genomes Project. The affected amino acid residues are highly phylogenetically conserved, from fungi (Neurospora crassa and Yarrowia lipolytica) to humans in the case of Arg211Cys, and from nematodes to humans in the case of Gly245Arg (figure 3). Finally, functional studies demonstrated a severe reduction in the steady-state levels of NDUFAF1 protein in Western blots of patient fibroblasts, with accumulation of abnormal complex I assembly intermediates on BN-PAGE analysis similar to those seen in the only other patient previously reported to have complex I deficiency caused by NDUFAF1 mutations.13 Our report confirms NDUFAF1 as a bona fide assembly factor of complex I, mutations of which cause isolated complex I deficiency.
Recent x-ray crystallographic studies have demonstrated the complex I holoenzyme to be an L-shaped macromolecule composed of four functional modules: the N module is responsible for NADH oxidation, the Q module is responsible for ubiquinone reduction, and the proximal PP and distal PD modules are responsible for proton translocation.9 10 Assembly of complex I has been extensively studied in the fungus N crassa and in human cells11 15 25 26; these studies, together with the phylogenetic profiling work of Pagliarini et al,7 have led to the identification of 24 putative complex I assembly factors (supplementary table 1 online). Sixteen of these have already been associated with human disease, including nine complex I deficiency syndromes and one mitochondrial disorder with multiple respiratory chain deficiencies.12 18 27 28 Interestingly, mutations in seven of these putative complex I assembly factors have been linked to non-mitochondrial diseases such as peroxisomal disorders, organic acidaemias and a ketone body utilisation defect (supplementary table 1 online). It is not clear whether reduced complex I activity may be involved in the pathogenesis of any of these disorders. While the precise mechanism of complex I assembly is still debated, a consensus view of a dynamic multidirectional process that includes the possibility of direct subunit exchange into pre-existing mature complex I has recently been proposed.11
NDUFAF1 was initially identified as the human homologue of CIA30, one of the first two complex I assembly factors identified in N crassa,25 and was demonstrated to be a ubiquitously expressed, mitochondrially targeted protein.20 21 NDUFAF1 is a protein of 327 amino acids and ∼37.7 kDa and contains no predicted transmembrane domains. Ryan et al29 described its N-terminally processed mitochondrial targeting signal, and Dunning et al13 showed that NDUFAF1 peripherally associates with the matrix face of the mitochondrial inner membrane. A role for NDUFAF1 in complex I assembly is also supported by RNA interference experiments, which showed disrupted complex I assembly in HeLa cells,21 and by recent studies of the testicular nuclear receptor (TR4)−/− knockout mouse model.30 TR4 is a member of the nuclear receptor superfamily of transcription factors and appears to regulate complex I activity by binding to hormone response elements in the NDUFAF1 promoter sequence. TR4−/− mice had a ragged-red fibre myopathy associated with complex I deficiency and reduced NDUFAF1 gene expression. Furthermore, lentiviral overexpression of NDUFAF1 was able to restore complex I activity and ATP generation to control levels in primary fibroblasts from TR4−/− mice.
The precise function of NDUFAF1 remains obscure; this is also true for all currently known complex I assembly factors. NDUFAF1 appears to be involved at an intermediate stage of complex I assembly, in contrast to C20orf7, NDUFAF3 and NDUFAF4, which are needed early in the assembly process, and to NDUFAF2, which has a role in the late stages.11 Tandem affinity purification experiments demonstrated that NDUFAF1 copurifies with two other putative complex I assembly factors, ECSIT and ACAD9.26 27 Interestingly, ACAD9 mutations have recently been reported to cause complex-I-deficient HCM, including a fatal infantile phenotype similar to that of our patient with NDUFAF1 mutations.19 27 Possible functions of the various putative complex I assembly factors/chaperones include iron–sulphur cluster synthesis (eg, NUBPL), translational coactivation of complex I subunits (as has been suggested for C20orf7 for ND1) and direction of nuclear-encoded complex I subunits to the correct intramitochondrial compartment (ie, to the matrix side of the enzyme or to the intermembrane space).11
Clinical recognition of mitochondrial heart disease can be difficult in the absence of multisystem features or a clear family history. The presence of lactic acidosis in a patient with heart failure usually reflects inadequate tissue oxygenation and perfusion rather than primary mitochondrial dysfunction, but persistence of lactic acidosis despite adequate ventilatory and circulatory support, as in the present case, should arouse suspicion of an underlying mitochondrial disorder. Echocardiographic features may also be helpful. Metabolic investigations in this infant were pursued because of the ventricular hypertrophy associated with poor systolic function, quite unlike the briskly contracting ventricles in HCM associated with sarcomeric protein abnormalities. There was also increased trabeculation suggestive of, but not definitive for, NCLV. The latter is a rare genetic cardiomyopathy that is characterised by hypertrabeculation of the left ventricle and appears to be an arrest of normal myocardial development.31 NCLV is frequently caused by mitochondrial disease, including mtDNA mutations and Barth syndrome.32 33
Mitochondrial cardiomyopathy is clinically and biochemically heterogeneous. Establishing a genetic diagnosis is challenging and achieved in only a minority of cases because of complex genetics, including bigenomic inheritance, and is guided by biochemical findings in biopsied tissue, which may show an isolated deficiency of one respiratory chain enzyme complex (most commonly complex I or complex IV) or a combined defect affecting multiple enzymes. In the case of cardiomyopathy, inaccessibility of the affected tissue poses further difficulties: endomyocardial biopsy has associated risks and does not provide sufficient tissue for conventional assays of mitochondrial respiratory chain function. For this reason, we and others use skeletal muscle for diagnostic purposes, accepting that this potentially lowers the diagnostic success rate since the mitochondrial biochemical defect may be limited to cardiac muscle.4 Demonstration of isolated complex I deficiency in the skeletal muscle from our patient led to screening of candidate genes. Complex I deficiency is genetically heterogeneous and may arise from mutations in the 45 structural subunits of the enzyme (including 7 mtDNA-encoded subunits and 38 nuclear-encoded subunits) or in any of the known and putative assembly factors.7 12 18 19 We first screened the entire mtDNA in our patient and subsequently performed systematic analysis of candidate nuclear genes previously reported to be associated with mitochondrial cardiomyopathy, eventually leading to the identification of compound heterozygous missense mutations in NDUFAF1.
The reported modes of inheritance of mitochondrial heart disease include autosomal recessive (representing the majority), X linked (eg, NDUFA1 and TAZ mutations), maternal (mtDNA point mutations) and sporadic (large-scale mtDNA rearrangements). A precise genetic diagnosis is important to provide parents with accurate prognostic and genetic counselling advice regarding recurrence risks and to guide future reproductive options. In this case, the parents had a successful pregnancy with donor egg in vitro fertilisation, resulting in a healthy child. Although subsequent demonstration of normal mtDNA sequence in the patient's skeletal muscle indicated that maternal inheritance was extremely unlikely, the parents opted for further donor egg in vitro fertilisation procedures, unfortunately without success. Identification of the underlying nuclear gene mutations allows these parents the option of prenatal or preimplantation genetic diagnosis for future pregnancies.
Cardiac muscle has a high energy requirement. Energy deficiency appears to be a common pathogenic mechanism in HCM: by directly affecting mitochondrial energy production (respiratory chain defects), by altering energy sensing (deficiency of AMP-activated kinase) or by increasing sarcomeric energy utilisation (sarcomeric protein mutations).3 However, cardiomyopathy is not an invariant feature of mitochondrial respiratory chain deficiency. For mtDNA-related disease, where mutations are present in multiple copies, this conundrum may be explained by tissue segregation and threshold effects, with a critical mutation load being required for biochemical expression. Other explanations are required for the phenotypic variation in nuclear gene defects causing mitochondrial disease34—for example, cardiomyopathy is not an invariant feature of mitochondrial disease caused by complex I assembly factor defects,12 suggesting differential importance of the various complex I assembly factors within the heart and possibly additional tissue-specific roles for these molecular chaperones. A relatively high expression of NDUFAF1 in cardiac tissues supports a specific role for this assembly factor in heart mitochondria.20 In addition, mitochondria play critical roles in intracellular calcium homeostasis, generation of reactive oxygen species (especially at complex I) and control of apoptosis; disruption of these functions is likely to contribute to HCM pathogenesis.
It is interesting to note that our case and the previously reported case with NDUFAF1 mutations had cardiomyopathy apparently triggered by viral illnesses, at 6 months in our case and at 15 months in the previous case.13 NDUFAF1 has recently been implicated in immune pathways, including T cell activation,35 36 and we speculate that this may explain why viral infection triggered decompensation and development of HCM in both patients with NDUFAF1 mutations. In marked contrast to our patient, the previously reported case showed normalised cardiac function, other than recurrent episodes of supraventricular tachycardia related to Wolff–Parkinson–White syndrome.13 His subsequent clinical course was characterized by multisystem problems, including developmental delay, mild cognitive impairment, cortical visual dysfunction, pigmentary retinopathy, kyphoscoliosis, osteoporosis and pubertal delay, with survival until the age of at least 21 years (age at time of report). Phenotypic heterogeneity is a phenomenon that is increasingly being recognised in nuclear-encoded mitochondrial disorders.34 37 Environmental factors, including age at exposure to first metabolic stress, severity of first metabolic stress (ie, viral infection) and autoimmune phenomena,38 may have contributed to the differential severity of the phenotypes in the two NDUFAF1-deficient cases. The reasons for the apparent reversal of cardiomyopathy in the case reported by Dunning et al are unclear. Improvement of mitochondrial disease symptoms has been reported with mutations in mitochondrial and nuclear genes, although the molecular mechanisms underlying this are not understood at present.39 40 The rapidly expanding knowledge of the molecular basis of mitochondrial disease will hopefully lead to a greater understanding of these issues in the future.
Acknowledgments
We are extremely grateful to the parents, who gave their consent and full support to this study.
References
Footnotes
Funding SR is supported by the Great Ormond Street Hospital Children's Charity. This project received grant funding from the Great Ormond Street Hospital/UCL Institute of Child Health Science Development Initiative and was made possible by a British–Italian partnership grant from the British Council.
Competing interests None.
Ethics approval Ethics approval was provided by the Great Ormond Street Hospital/Institute of Child Health research ethics committee.
Provenance and peer review Not commissioned; externally peer reviewed.