Article Text

Statistics from Altmetric.com
Five patients with campomelic dysplasia who have survived (age range 7 to 20 years) are described, all of whom have molecular or cytogenetic evidence of campomelic dysplasia. The phenotype and radiological features of these cases are consistent. Complications in this group include recurrent apnoea and upper respiratory infections, progressive kyphoscoliosis, mild to moderate learning difficulties, short stature, and dislocation of the hips. All five had very similar facial features. The radiological features include hypoplastic scapulae, defective ischiopubic ossification, absent or hypoplastic patellae, and spinal dysraphism.
Campomelic dysplasia (CMD) is a rare skeletal dysplasia resulting from mutations in SOX9. It is usually lethal in the first year of life. Three-quarters of the cases with a male karyotype have complete or partial sex reversal.1 The skeletal changes in the neonatal period are well recognised and include hypoplastic scapulae, bowing of the long bones, vertical narrow iliac bones, and absence of ossification of the thoracic pedicles.
The case histories of five children who share a number of clinical and radiological features are presented.
CASE REPORTS
Case 1
A mother was diagnosed at the age of 18 years after giving birth to a daughter with the classical features of campomelic dysplasia.2
The daughter had shortening of all four limbs, tibial bowing, with skin dimpling over the apex of each tibia. There was bilateral talipes equinovarus and relative macrocephaly (head circumference on the 50th centile, length <3rd centile). She had micrognathia and a depressed nasal bridge. The karyotype was normal female. She had the classical radiological features of campomelic dysplasia with hypoplastic scapulae and absent pedicles in the mid-thoracic region, 11 pairs of ribs, shortening of the long bones with bowing, narrow iliac bones with dislocation of the hips, short first metacarpals, and short phalanges in the hands and feet.2 She died of respiratory problems in the neonatal period.
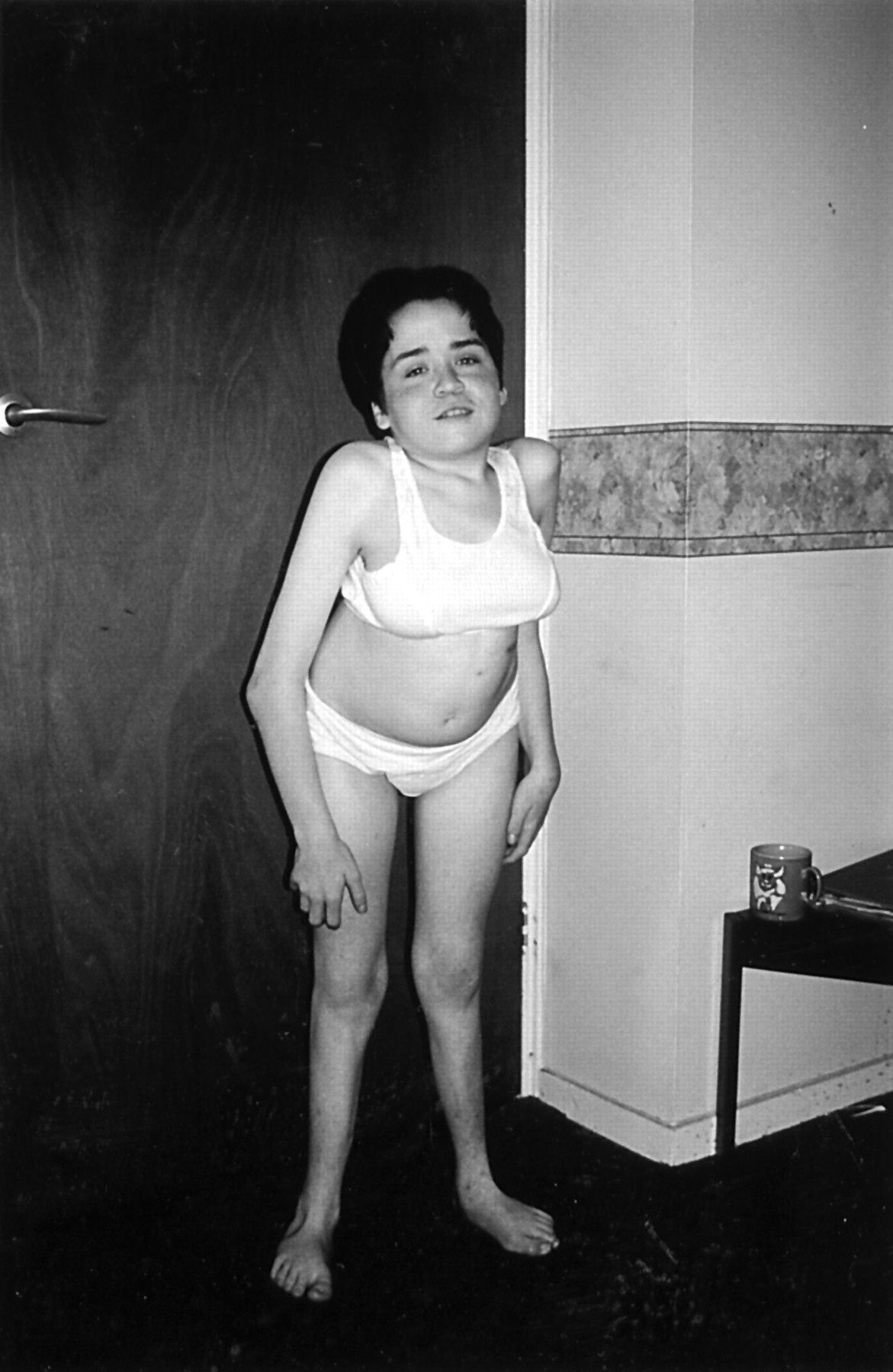
At that time her mother was examined and found to have some skeletal abnormalities. She was below the 3rd centile for height (1.5 m) with a flat face, long philtrum, depressed nasal bridge, small mouth, and micrognathia (fig 1). A right fibula osteotomy was required at the age of 11 years because of a 4 cm asymmetry of her lower limbs. The halluces on both sides were short. Both thumbs and little fingers were short and the nails were hypoplastic. She had conductive hearing loss of the left ear and was myopic. Intelligence was normal but seizures began at the age of 13 requiring treatment with sodium valproate. Radiographs showed a small left iliac bone with non-ossification of the inferior pubic rami and short ischia on both sides, hypoplastic scapulae, 11 pairs of ribs, short first metacarpals, short middle phalanges of the index and little fingers, and thoracic scoliosis. The patellae were small and there was mild bowing of the tibia (see fig 1A and B in Lynch et al2 and Offiah et al3).

Patient 1 aged 18 years.
Mutation analysis of SOX9 confirmed deletion at C155 resulting in a frameshift and truncation of the product at amino acid 59 (A J Schafer, personal communication). The SOX9 mutation was found in the mother and daughter.
Case 2
This affected boy is currently 11 years old. He was born to Pakistani consanguineous parents (half first cousins) who had a termination of pregnancy for a fetus which was also affected by campomelic dysplasia. He has one normal older sister and a brother who died at 1 day of age from congenital heart disease.
This baby was born at 37 weeks’ gestation by caesarean section for previous sections. He weighed 2400 g at birth. Immediate resuscitation by intubation was required for respiratory distress.
Clinical features included short long bones and marked bilateral talipes equinovarus. He was dysmorphic (fig 2) with macrocephaly, hypertelorism, depressed nasal bridge, low set ears, micrognathia, and a posterior submucous cleft of the palate. He had marked tracheomalacia and recurrent apnoea and required a tracheostomy until the age of 6 years. He also required a gastrostomy for poor feeding.

Patient 2 aged 3.5 years.
At 3½12 years, his height was 92 cm (<3rd centile) and head circumference was 54 cm (>97th centile). There was moderate global developmental delay. His gross motor development was very delayed and he did not walk until he was 3½12 years old. Hearing was impaired but vision was normal. There was marked joint laxity and a progressive, moderate kyphoscoliosis, dislocation of one hip, and unstable knees.
Radiographs showed a double curve scoliosis with lumbar spine dysraphism (fig 3) and an exaggerated lumbar lordosis with short pedicles and pronounced scalloping of the lumbar vertebral bodies. Both scapulae were hypoplastic (fig 4). There was also defective ischiopubic ossification, narrow iliac bones, and hypoplastic patellae (fig 2A and fig 1C, Offiah et al3).

Case 2: AP thoracolumbar spine (aged 9 years) showing double curve scoliosis and lumbar spinal dysraphism.
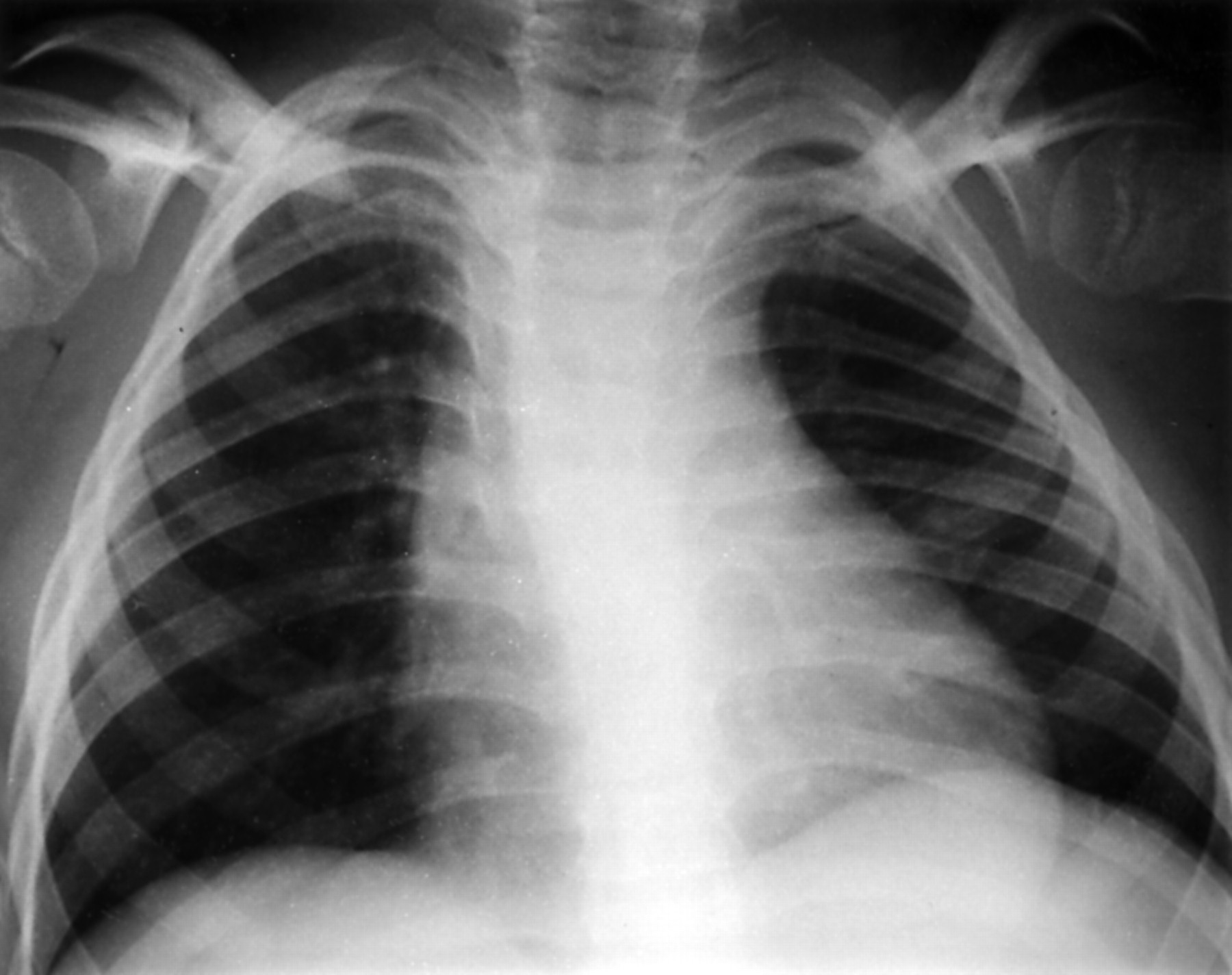
Case 2: AP chest (aged 7.5 years) showing hypoplastic scapulae with absent scapular wings and lateral clavicular hooks.
A SOX9 mutation (H65Y) has been found in this case which has previously been shown greatly to reduce the DNA binding activity of the SOX9 protein (McDowall et al,4 patient 10).
Case 3
This child is now 7 years old but was reviewed at the age of 1 year. She was born at 42+ weeks by normal vaginal delivery to unrelated parents who had a previous normal child. The pregnancy had been uneventful apart from hyperemesis and antenatal scans were reported as normal at 19 and 41 weeks. At birth, intubation and ventilation were required immediately but she was successfully extubated after 24 hours. She required a nasopharyngeal tube for five weeks and there was marked hypotonia.
She had a variable degree of stridor and recurrent apnoea usually associated with feeding. There was a progressive kyphoscoliosis from the age of 3 months requiring spinal fusion and brace. Her hips were not dislocated but she had dislocatable knees. There was mild conductive hearing loss but vision at one year was normal. There was mild global developmental delay but gross motor skills were more markedly delayed. On examination, she was short with a height of 68 cm (<3rd centile) at 11 months, but not asymmetrical. She was dysmorphic with macrocephaly (98th centile), depressed nasal bridge, hypertelorism, epicanthic folds, micrognathia, and a cleft of her soft palate. There was a significant pectus carinatum and bilateral metatarsus varus. She had some bowing of the long bones in her lower limbs with bilateral pretibial dimples. There was complete sex reversal with normal female external genitalia.
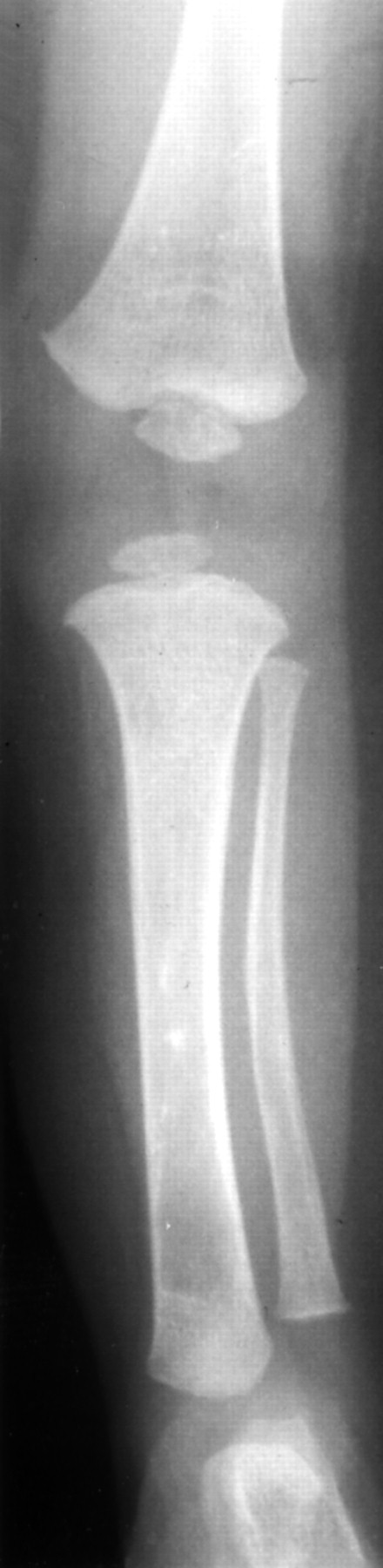
Radiographic findings include defective ischiopubic ossification (fig 2B, Offiah et al3), narrow iliac bones, anterior bowing of the tibia, disproportionately short fibulae, and hypoplastic distal tibial epiphyses (fig 5). There was a double curve scoliosis with segmentation defects of the mid-dorsal spine, narrow iliac bones, and hypoplastic scapulae.

Case 3: lateral tibia and fibula (aged 1 year) showing disproportionately short fibula, hypoplastic distal tibial epiphysis, and anterior bowing of the tibia.
Chromosome analysis showed a male karyotype with a de novo paracentric inversion of chromosome 17 (46,XY,inv(17)(q11.2,q24.3-q25.1).
Case 4
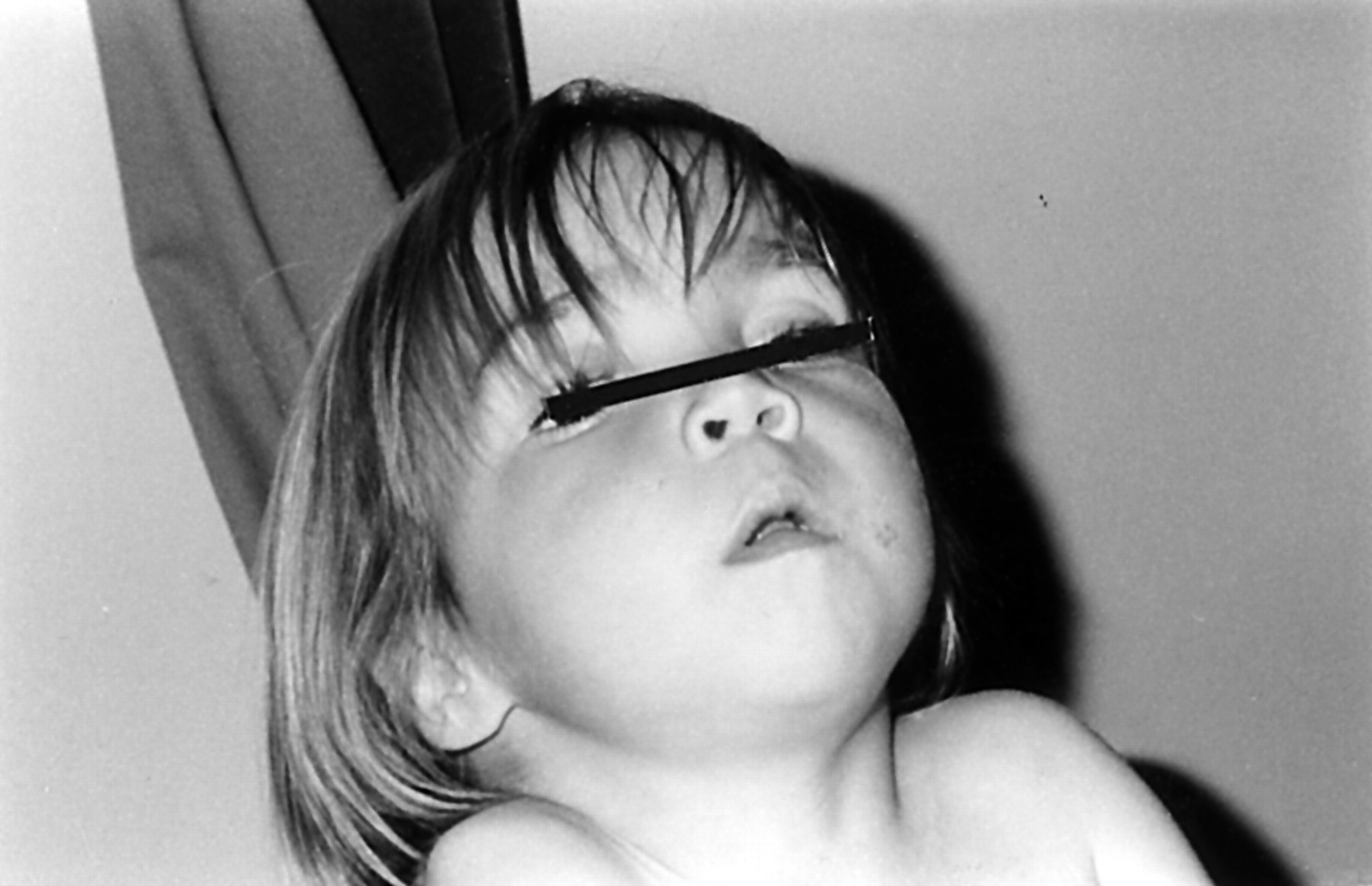
This 11 year old girl was the first child of healthy, unrelated parents. She was born by emergency caesarean section for failure to progress at 42 weeks’ gestation. During the neonatal period her breathing was noted to be noisy and she required observation but no ventilation.
On examination at the age of 4 years, she was dysmorphic (fig 6) with relative macrocephaly (between the 50th and 90th centile), frontal bossing, depressed nasal bridge, hypertelorism, micrognathia, and low set ears. There was disproportionate short stature (<3rd centile) with a short trunk and relatively long limbs.

Patient 4.
She had a severe and progressive kyphoscoliosis for which she has had unsuccessful corrective surgery. There have been recurrent upper and lower respiratory infections and persistent mild stridor. There was mild developmental delay, particularly affecting gross motor development.
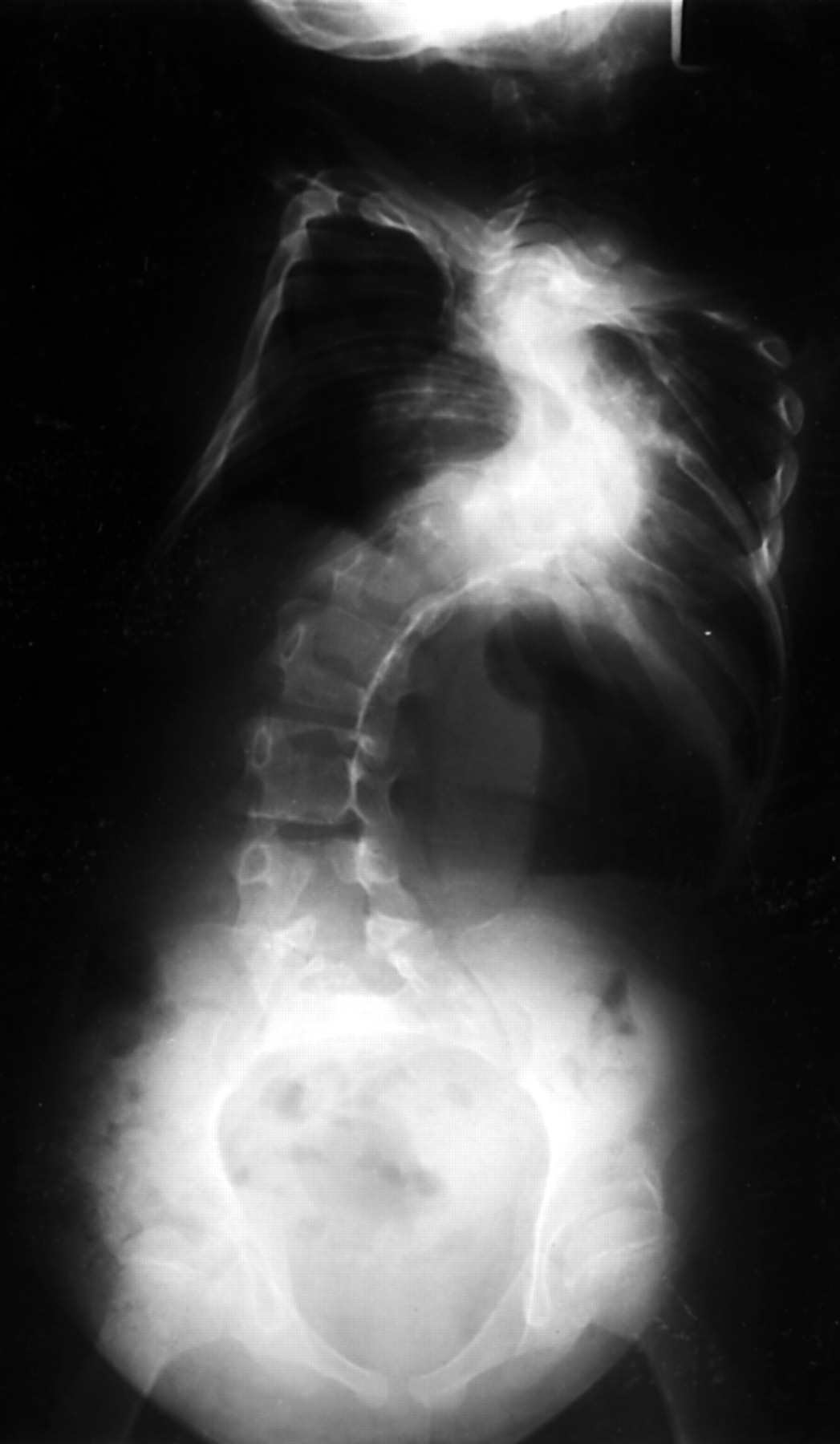
The radiological findings showed marked kyphoscoliosis, with spina bifida of the lower cervical and mid-thoracic spine. There were hypoplastic scapulae (fig 7) and bilateral dislocation of the radial heads. There was defective ossification of the inferior pubic rami with dislocation of the left hip and narrow, vertical iliac wings (fig 8).

Case 4: AP chest (aged 5.5 years) showing spina bifida of the lower cervical spine, hypoplastic scapular wings, long acromian processes, and a thoracic scoliosis concave to the left.

Case 4: AP pelvis (aged 9 years) showing short ischia, defective ossification of the inferior pubic rami, dislocated left hip, narrow vertical iliac wings, and slender superior pubic rami.
Chromosome analysis showed a female karyotype but with a de novo balanced translocation, 46,XX, t(4;17)(q21.3;q23.3).
Case 5
This woman is now 21 years old (she has four normal sibs). Birth was at term by emergency caesarean section for fetal distress. She had respiratory distress in the neonatal period but was not ventilated. At birth, she was noted to be short limbed and had bilateral pretibial dimples. There was bilateral talipes equinovarus which was more severe on the right.
She was dysmorphic with a depressed nasal bridge, hypertelorism, micrognathia, and low set ears (fig 9). There was a high arched palate but no cleft palate. On examination, her height was 119 cm (<<3rd centile) with a relatively large head circumference of 53 cm (10th centile). She had disproportionate short stature, with a short trunk and relatively long limbs. Supination of her forearms was restricted owing to bilateral dislocation of the radial heads. The left hand had long index and middle fingers but the ring and little fingers were short with contractures at the distal interphalangeal phalanges. There was bilateral hallux valgus and pes planus.

Patient 5 aged 21 years.
She had many complications with recurrent apnoea and chest infections. The apnoea improved following adenoidectomy at the age of 2 years. A severe, progressive kyphoscoliosis developed from the age of 9 months. This required two major but unsuccessful operations. Conductive hearing loss required grommets and hearing aids and there was myopia. There was marked dental caries and irregular teeth. She had moderate learning difficulties and attended a special needs school.
The radiological features included bowing of the long bones and delayed ossification of the epiphyses (fig 10), short first metacarpals with short middle and distal phalanges, and dislocated radial heads. There was defective ischiopubic ossification (fig 2D, Offiah et al3) and small patellae. The spine showed a significant scoliosis with cervical and lumbar dysraphism (fig 11).
Case 5: AP left tibia and fibula (aged 2 years) showing a short fibula, bowing of the tibia and fibula, small distal femoral and proximal tibial epiphyses, and delayed ossification of the distal tibial and fibular epiphyses.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Case 5: AP thoracolumbar spine (aged 11 years) showing significant scoliosis, cervical and lumbar spinal dysraphism, defective ischiopubic ossification, and “drumstick” proximal femora.
The karyotype was normal female and SOX9 mutation analysis showed a de novo cytidine to thymidine transition at nucleotide 509, which results in substitution of proline 170 with leucine. This mutation has not been previously reported.
DISCUSSION
Campomelic dysplasia is generally considered to be a lethal skeletal dysplasia. However, it is clear from published reports that a small proportion of affected subjects survive. In a previously reported series of 36 patients, there were three survivors, giving a survival rate of between 5% and 10%.1 However, it is possible that some surviving patients with a milder phenotype are not diagnosed, so the survival rate may be higher.
The dysmorphic features of all five cases are consistent and distinctive (table 1). Characteristically there is relative macrocephaly, depressed nasal bridge, hypertelorism, long philtrum, micrognathia with or without a cleft palate, and hypotonia.
A comparison of the clinical facial features and complications in the five patients presented
The main clinical problems in the neonatal period are respiratory. These include recurrent apnoea, chest infections, and stridor, sometimes requiring tracheostomy. Later complications include conductive hearing loss, developmental delay, (especially gross motor), and dental caries with irregular teeth. Myopia was present in the two oldest patients (cases 1 and 5) and may be a complication in later life.
Other complications have been reviewed in orthopaedic publications.5–7 These reports emphasise the presence of a severe and progressive kyphoscoliosis associated with increased morbidity. Other orthopaedic problems include talipes equinovarus, congenital subluxation or dislocation of the hips, and dislocation of the radial heads causing limitation in supination.
There is a striking change in the disproportion of the short stature with age. At birth there are short limbs with normal length of the trunk, but with the progression of the kyphoscoliosis the trunk becomes relatively shorter than the limbs (fig 9).
Sex reversal was only present in one of these cases (case 3) and was associated with a paracentric inversion of the long arm of chromosome 17. The other male in this series was not sex reversed (case 2). In a previously reported series, 73% of patients with a male karyotype had female or ambiguous genitalia. Only four patients had a male karyotype with normal male genitalia. One of these cases is described here, another survived for four months, and two were terminated in pregnancy because of a prenatal diagnosis of a lethal skeletal dysplasia. From these small numbers it is impossible to postulate that sex reversal is associated with a more severe phenotype.
There are two possible explanations for the survival of these patients with CMD. The first explanation is mosaicism of the SOX9 mutation. This is the most likely reason for survival in case 1. This woman had clinical and radiological evidence of asymmetry and gave birth to a girl with classical lethal CMD. The father of case 2 was also shown to carry the same SOX9 mutation as his affected son but was clinically unaffected.4 He also had a further fetus affected by CMD. It is likely that he is also mosaic for the mutation.
Secondly, chromosomal rearrangements involving chromosome 17q have been shown to cause CMD without disrupting the SOX9 gene. Two of the patients reported here had a de novo chromosomal translocation involving chromosome 17q. A previous study described three patients with campomelic dysplasia and a chromosomal translocation (involving 17q); two of these patients were 2.5 years old and one was still alive at the age of 26 years.8 These rearrangements may somehow reduce but not abolish the expression of the gene resulting in a milder phenotype.9,10 Two of these patients had sex reversal, again suggesting that this feature is not a significant feature in terms of prognosis in this condition.
The SOX9 protein has been shown directly to regulate the COL2A1 (type II collagen) gene.11,12 It is also a transcription factor that is essential for chondrocyte differentiation and cartilage formation.13,14 This may explain the overlapping features with spondyloepiphyseal dysplasia congenita, Stickler syndrome, and Kniest syndrome, such as facial dysmorphism (depressed nasal bridge, micrognathia, cleft palate), epiphyseal, and spinal involvement.
There is considerable radiological overlap with ischiopubic-patella syndrome (IPP).3 This suggests that some of the cases reported as the more severe form of IPP may be surviving campomelic dysplasia. The radiological features in these five patients are characteristic and described in detail by Offiah et al.3 All five of the patients described here have the characteristic radiological features of IPP but have genetic or cytogenetic evidence of CMD. We believe that the IPP syndrome is a heterogeneous group. The more severe cases are probably survivors of CMD and may have mosaic or mild mutations in SOX9 or cytogenetic rearrangements of chromosome 17. The milder forms of IPP with radiological changes confined to the pelvis and patella are a separate entity and are probably not related to SOX9, although further studies on these patients would be required to prove this. We suggest that this “mild” group is called “small patella syndrome”, as originally described.15
Surviving CMD is a distinct and recognisable entity; the complications appear to be consistent within this group and overlap with the type II collagenopathies.
Acknowledgments
We would like to thank Dr Christine Garrett, Kennedy-Galton Centre, Northwick Park Hospital, Harrow, London, and Dr Sally Lynch, Newcastle, for their contribution.