

Abstract
Alzheimer’s disease (AD) has a multifactorial etiology, which requires a single multi-target approach for an efficient treatment. We have focused on E2F4, a transcription factor that regulates cell quiescence and tissue homeostasis, controls gene networks affected in AD, and is upregulated in the brain of Alzheimer’s patients and of APP/PS1 and 5xFAD transgenic mice. E2F4 contains an evolutionarily-conserved Thr-motif that, when phosphorylated, modulates its activity, thus constituting a potential target for intervention. Here we show that neuronal expression in 5xFAD mice of a dominant negative form of E2F4 lacking this Thr-motif (E2F4DN) potentiates a transcriptional program consistent with global brain homeostasis. The latter correlates with attenuation of both microglial immune response and astrogliosis, modulation of Aβ proteostasis, and blockade of neuronal tetraploidization. Moreover, E2F4DN prevents cognitive impairment and body weight loss, a known somatic alteration associated with AD. We propose E2F4DN-based gene therapy as a promising multifactorial approach against AD.
Introduction
Alzheimer’s disease (AD) is characterized by progressive neurodegeneration that leads to cognitive impairment and eventually to dementia, in association with somatic alterations including body weight loss (1). Two main neuropathological hallmarks, derived from altered proteostasis, can be found in the brain of AD patients, namely senile plaques containing β-amyloid peptide (Aβ), which derives from amyloid precursor protein (APP) processing, and intraneuronally-located neurofibrillary tangles (NFTs), constituted of hyperphosphorylated tau protein (2). Compelling evidence indicates that AD has a multifactorial etiology (3), with neuroinflammation as a relevant central mechanism (4), due to its capacity to exacerbate Aβ and tau pathologies (5). Other early-onset processes that cooperate in the etiology of AD include synapse loss (6), altered glucose metabolism (7), oxidative stress (8), chronic hypoperfusion (9), and neuronal cell cycle re-entry (10), the latter leading to neuronal tetraploidization (NT) (11). These processes interact with each other resulting in synergistic effects. For instance, neuronal cell cycle re-entry can induce NFTs, extracellular deposits of Aβ, gliosis, synaptic dysfunction and delayed neuronal cell death, which all together lead to cognitive deficits [reviewed by (12)]. Additionally, oxidative stress affects synaptic activity and triggers abnormal cellular metabolism that in turn could affect the production and accumulation of Aβ and hyperphosphorylated tau (8). Cell cycle-reentry can also cooperate with altered glucose metabolism in the etiology of AD (13), and synapse dysfunction might also underlie the AD etiology (14). Mutual interaction of all these etiological factors makes it difficult to appropriately target the disease and, to date, no effective therapies against AD are available. This is likely due to the monospecific nature of most drugs that have been tested so far. Therefore, a paradigm shift is necessary and the design of a single multi-target approach against this complex disease is mandatory (3).
A recent study has proposed E2F4 as a major regulator of most AD-specific gene networks (15), and other bioinformatics-based studies suggest that E2F4 participates in this disease (16-18). Moreover, a genome-wide association study for late onset AD has identified a single nucleotide polymorphism that modifies a DNA binding motif of E2F4 as relevant for the disease (19). E2F4 can potentially regulate over 7,000 genes involved in several AD-affected processes, including its well-known cell cycle regulation function, as well as DNA repair, RNA processing, stress response, apoptosis, ubiquitination, protein transport and targeting, protein folding, and I-κB kinase/NF-κB cascade (20). In addition, E2F4 can bind to the promoters of 780 transcription factors, suggesting that E2F4 can regulate broad classes of genes indirectly (20), either through Rb family-dependent or independent mechanisms (21). Supplementary data reported by this latter study indicate that E2F4 can physically interact with relevant synaptic regulators including fragile X mental retardation 1 (FMR1), Fragile X Mental Retardation Syndrome-Related Protein 1 (FXR1), and FXR2, as well as with IQ motif and Sec7 domain-containing protein 2 (IQSEC2). E2F4 can also interact with subunit 2 of biogenesis of lysosomal organelles complex-1 (BLOC-1), BLOC-1-related complex subunit 5, and SNARE-associated protein snapin (21), which are crucial for intracellular vesicle trafficking and synaptic vesicle recycling (22). Other E2F4 interactors described by (21) are PCMT1/PIMT, an enzyme that repairs abnormal L-isoaspartyl linkages in age-damaged proteins (23), as well as Prefoldin 1 (PFDN1) and PFDN4, two members of a molecular chaperone complex that plays an important role in proteostasis and contributes to the formation of non-toxic Aβ aggregates in vitro (24). Therefore, E2F4 could fulfill AD-relevant functions besides those linked to its DNA binding activity.
E2F4 is a phosphoprotein (21), and previous studies from our laboratory have shown that E2F4 can be phosphorylated by p38MAPK (25), a major stress kinase that is activated in AD (26). In the chick, this phosphorylation takes place at the Thr261/Thr263 motif, orthologous of Thr249/Thr251 in mouse E2F4 and Thr248/Thr250 in human E2F4 (25). Accordingly, a recent study has identified Thr248 (Thr249 in mouse E2F4) as a major phosphorylatable Thr residue in E2F4 (21). E2F4 expression is upregulated in cortical neurons from APPswe/PS1dE9 (APP/PS1) mice, a known AD mouse model, in association with phosphoThr immunoreactivity (27). Remarkably, a similar E2F4 upregulation can also be observed in the AD prefrontal cortex (10) and in human neurons derived from FAD patient-specific hiPSCs (18), as well as in cortical neurons from 5xFAD mice (27), another mouse model of AD that expresses human APP and PS1 containing five pathological mutations (28).
We have demonstrated that a phosphomimetic form of chick E2F4 with Thr261Glu/Thr263Glu mutations leads to cell cycle re-entry in differentiating chick neurons lacking p38MAPK activity, while a dominant negative form of chick E2F4 (E2F4DN) containing Thr261Ala/Thr263Ala substitutions blocks NGF-induced cell cycle re-entry in these cells (25). This indicates that the phosphorylation of the Thr conserved motif alters the normal functioning of E2F4 as a quiescent regulator, a process that could participate in the etiology of AD. This phosphorylation might also alter other homeostatic processes regulated by E2F4, and if so E2F4DN could be a potential therapeutic tool for this disease.
In this study, we generated a knock-in mouse strain expressing mouse E2F4DN in neurons, which were mated to 5xFAD mice. We show that neuron-specific expression of E2F4DN in 5xFAD mice prevents NT and induces a transcriptional program that attenuates the immune response, reduces the stability, aggregation and toxicity of Aβ, and includes markers of synapse formation, improved glucose metabolism and vascular integrity, as well as of decreased oxidative stress, glycophagy and cell starvation. Although the attenuated neuroinflammatory response observed in 5xFAD/E2F4DN mice correlates with the presence of enlarged Aβ deposits, the presence of the latter does not disturb cognition, as occurs in asymptomatic AD individuals (29). Indeed, neuronal E2F4DN restores cognitive function and, surprisingly, prevents AD-associated somatic alterations such as body weight loss. Therefore, we propose E2F4DN as a promising multifactorial, therapeutic agent against AD.
Results
Immune response modulation in 5xFAD mice expressing E2F4DN
We generated an E2F4DN knock-in mouse strain (E2F4DN mice) by inserting the coding sequence of mouse E2F4 containing Thr249Ala/Thr251Ala mutations, Myc tagged at the C-terminus, into the gene encoding the microtubule-associated protein tau, as previously described by (30) for EGFP knock-in mice (EGFP mice) (Fig. 1a). This construct was germ-line transmitted to the progeny (Fig. 1d) and, as expected, neurons from E2F4DN mice expressed the E2F4DN-myc protein, as evidenced by NeuN/Myc-specific immunohistochemistry (Fig. 1b,c). Western blot confirmed the specific expression of E2F4DN-myc in the cerebral cortex of these mice (Fig. 1e). As previously reported for EGFP mice (30), homozygous E2F4DN mice were viable and fertile.

(A) Scheme showing the DNA construct used to generate the E2F4DN mice. Red: Mapt Exon I containing the NcoI restriction site where the E2f4dn-myc cassette was inserted. pA: Pgk-1 polyadenylation signal, Neo R: Pgk-Neor sequence. Primers #1 and #2, for the 5’ flanking region, are shown in blue while primers #3 and #4, for the 3’ flanking region, are depicted in red (see Methods for details). (B) A cerebral cortex cryosection from WT mice immunostained with anti-NeuN (red) and anti-Myc tag (green), and counterstained with DAPI. (C) Cerebral cortex cryosections from E2F4DN mice immunostained with anti-NeuN (red) and anti-Myc tag (green), and counterstained with DAPI. (D) Genomic DNA from control R1 cells (Contr.), clone #174, and three mice descendants from a cross between the founding chimera and a C57BL/6J mouse, amplified with primers #1 and #2. Arrow: band specific for the E2F4DN construct. (E) Western blot performed with extracts from cerebral cortex of WT and E2F4DN mice using an antibody against c-Myc tag. Loading control performed with an anti α-tubulin. Asterisk: unspecific band. Arrows: specific bands. Scale bar: 20 μm (B, C upper panels), 80 μm (C bottom panels).
To identify transcriptional changes induced by neuronal E2F4DN in AD, we crossed 5xFAD mice with both E2F4DN or control EGFP mice. We then performed RNA-seq analysis using total RNA isolated from the cerebral cortex of 5xFAD/EGFP and 5xFAD/E2F4DN mice at 3 month of age, an early stage in which incipient AD-associated neuropathology is already evident (31). This analysis indicated that, in addition to the transgenes, 273 genes were differentially expressed between both genotypes, 71 of which have been documented to participate in AD (Table S1). E2f4 was 2.4-fold enriched in 5xFAD/E2F4DN mice, indicating that the E2F4DN transgene is expressed at physiological levels.
We compared the differentially-expressed genes encoding characterized proteins (248 in total) with those genes whose expression is modulated in the cerebral cortex of APP/PS2 mice (32), another related murine model of Alzheimer. Consistent with the important role of neuroinflammation in AD (4), Srinivasan et al. (32) found that 84 genes plus an unprocessed pseudogene, were upregulated in APP/PS2 mice (74 of microglial origin) (Table S2). 36 of these genes were also upregulated in the cerebral cortex of 5xFAD/E2F4DN mice (Table S2). We confirmed by quantitative polymerase chain reaction (qPCR) the upregulation of a selected subset of these genes, including C4b, Cd84, Lgals3bp, Mpeg1, and Slc11a1 (Fig. 2a). Functional GO term annotation indicated that the microglia-expressed genes common to 5xFAD/E2F4DN and APP/PS2 mice are mostly involved in innate immune response (Common Genes; Table S3). In contrast, the microglia-specific genes unique in APP/PS2 mice (Table S2) were mainly involved in positive regulation of cytokine production (APP_PS2 Unique Genes; Table S3). This suggests that neuronal E2F4DN potentiates a non-cytotoxic immune response in the cerebral cortex of 5xFAD mice.

They include genes modulated in the cerebral cortex of APP/PS2 mice (A), strongly upregulated DAM genes (B), low or non-upregulated DAM genes (C), phagocytosis-related genes (D), inflammation-related genes (E), and Gfap, an astrocyte marker (F). Relative gene expression was normalized to Rps18 rRNA levels and expressed as 2ΔCt (obtained values were adjusted by the factor indicated between brackets). *p<0.05; **p<0.01; ***p<0.001 (Unbalanced two-way ANOVA, followed by post hoc Student’s t test).
The potentiation of an attenuated immune response by E2F4DN was further confirmed by browsing the MGI Mammalian Phenotype database against the protein-encoding genes modulated by E2F4DN in the cerebral cortex of 5xFAD mice that are absent in the study by (32) (212 in total). Decreased interferon-gamma secretion and abnormal immune system physiology were the two major phenotypes obtained, while decreased CD4-positive, alpha beta T cell number, decreased interleukin-12 secretion, abnormal T cell activation, and abnormal cytokine secretion were found among other prominent phenotypes (MGI Mammalian Phenotype; Table S3).
To further explore the effect of E2F4DN on the immune system, we focused on the molecular signature specific of disease-associated microglia (DAM) (33), recently renamed as activated response microglia (4). This signature can be modulated to either proinflammatory or anti-inflammatory DAM states (34). In the cerebral cortex of 5xFAD mice, the transition from homeostatic microglia to the DAM population is a gradual process, initiated by 3-4 months of age (31, 33). RNA-seq analysis indicated that neuronal E2F4DN accelerated this process in the cerebral cortex of 5xFAD mice since several DAM-specific genes were upregulated by E2F4DN, but not by EGFP (Fig. 2b). In contrast, and consistent with the accelerated expression of immune genes in the hippocampus of 5xFAD mouse (31), a significant increase of microglial markers, including both DAM-specific (Fig. S1a) and innate immune response (Fig. S1b) genes, was apparent in the hippocampus of 3 month-old 5xFAD/EGFP mice. As in the cerebral cortex, neuronal E2F4DN expression potentiated the expression of the immune-specific genes in this latter tissue (Fig. S1a,b).
The DAM-specific molecular signature upregulated in the cerebral cortex of 5xFAD/E2F4DN mice included the stage 1 DAM genes Tyrobp, Ctsd, and Lyz2, and the stage 2 DAM genes Trem2, Cst7, Ccl6, Itgax, Clec7a, and Lilrb4. (33). Other DAM genes described by these latter authors were not upregulated, suggesting that the presence of E2F4DN leads to a distinct DAM-associated phenotype. qPCR amplification of a representative subset of upregulated DAM genes (Tyrobp, Trem2, Cst7, Ccl6, Itgax, and Clec7a) confirmed the potentiation of the DAM-like signature in the cerebral cortex of 5xFAD/E2F4DN mice (Fig. 2b). Other DAM-specific genes not significantly detected in the RNA-seq analysis were either not upregulated (Axl) or upregulated at quite low levels (Csf1 and Apoe) when analyzed by qPCR (Fig. 2c).
AXL is a receptor tyrosine kinase crucial for the recognition of phosphoserine moieties in both apoptotic bodies and synapses to be pruned, and the subsequent activation of phagocytic capacity of microglia (35). Therefore, the lack of Axl upregulation is consistent with reduced phagocytic capacity of microglia in the cerebral cortex of 5xFAD/E2F4DN mice. The upregulation of Anxa5 expression by E2F4DN (Fig. 2d) also supports the reduced phagocytic capacity of microglia in the cerebral cortex of 5xFAD/E2F4DN mice. Anxa5 encodes Annexin V, a cytosolic protein that can suppress phagocytosis when extracellularly located due to its capacity to interact with membrane phospholipids (36). The slight increase of Csf1 in the cerebral cortex of 5xFAD/E2F4DN mice (Fig. 2c) is consistent with the attenuation of the inflammatory phenotype since colony stimulating factor 1, the Csf1-encoded protein, is a cytokine that stimulates phagocytic, cytotoxic and chemotactic activity in macrophages (37). Another relevant DAM-specific gene found to be upregulated at quite low levels is Apoe (Fig. 2c). Therefore, the capacity of ApoE to prevent the expression of the anti-inflammatory factor TGFβ and to favor cytotoxicity by microglial cells (38) as well as to potentiate the activated response to Aβ (4) seems to be attenuated in the DAM-like phenotype observed in 5xFAD/E2F4DN mice. Accordingly, we found that a number of pro-inflammatory DAM cell markers, including Ptgs2, Il1b, Il12b, Cd44, Kcna3, Nfkb1, Stat1 and Rela (39), were not significantly upregulated in the cerebral cortex of 5xFAD/E2F4DN mice (Table S1), a finding that was confirmed by qPCR using Il1b as a representative pro-inflammatory gene (Fig. 2e). Moreover, the TNFα-specific gene Tnfa was not significantly upregulated in the cerebral cortex of 5xFAD/E2F4DN mice either (Fig. 2e), further supporting that the inflammatory potential of the E2F4DN-dependent DAM-like phenotype is weakened. qPCR also confirmed the upregulation in 5xFAD/E2F4DN mice of other relevant genes with capacity to attenuate the inflammatory response. They include Serpina3n and Spp1 (Fig. 2e). The former encodes a Granzyme B inhibitor that induces neuroprotection (40), while the latter encodes osteopontin (OPN), a tissue repair gene known to regulate immune cell function and to respond to brain injury (4). OPN also modulates the ability of macrophage to resist pathogenic forms of Aβ (41). In addition, Pglyrp1, which encodes a peptidoglycan recognition protein that is expressed in polymorphonuclear leukocytes and is involved in antibacterial immunity and inflammation (42), is downregulated by E2F4DN (Fig. 2e). Finally, Tgfb, which encodes the anti-inflammatory cytokine TGFβ1 (38), was slightly upregulated in the cerebral cortex of 5xFAD/E2F4DN mice (Fig. 2e).
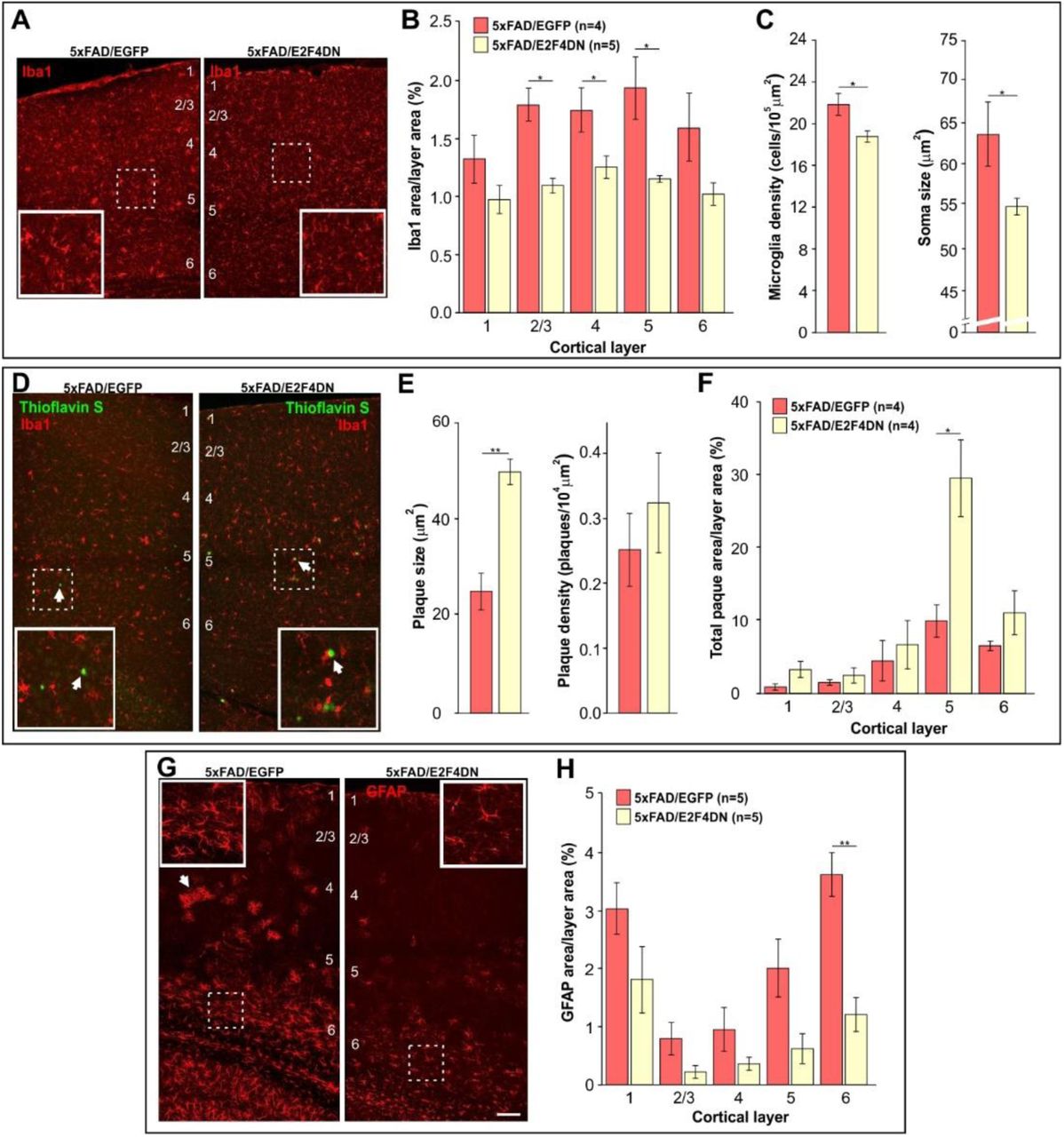
To confirm that neuronal expression of E2F4DN favors an attenuated microglial response in the cerebral cortex of 5xFAD mice, we immunolabeled cortical sections of 5xFAD/EGFP and 5xFAD/E2F4DN mice of 3 months of age with the specific microglia marker Iba1. This analysis demonstrated that the presence of E2F4DN significantly diminished the area occupied by microglial cells (Fig. 3a), mostly in layers 2-5 (Fig. 3b). A reduction of the area occupied by Iba1 immunoreactivity was also evident in the hippocampus of 5xFAD/E2F4DN mice (Fig. S2a,b). The attenuated microglial response in 5xFAD mice expressing E2F4DN was further evidenced by morphological criteria (43) since both the density and the size of cell somas harboring Iba1-specific immunoreactivity was significantly reduced in the cerebral cortex of 5xFAD/E2F4DN mice as compared with 5xFAD/EGFP mice (Fig. 3c).
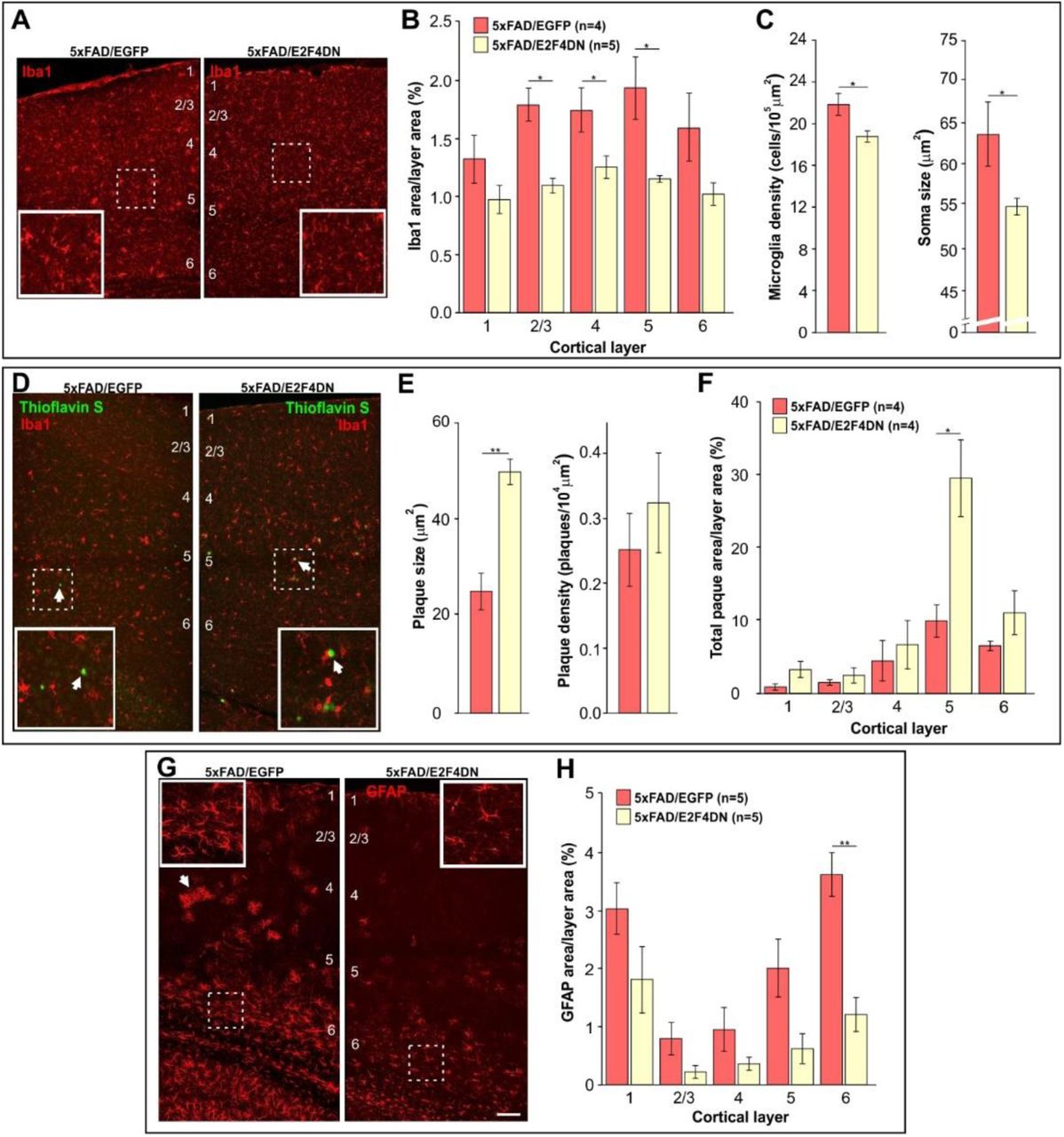
(A) Iba1 immunostaining in the cerebral cortex of mice of the indicated genotypes. (B) Percentages of the areas occupied by Iba1 immunostaining in the indicated cortical layers. (C) Density and size of Iba1-positive soma, evaluated as described by (43). (D) Dense core plaques (Thioflavin S labeling, arrows) co-stained with Iba1 in the cerebral cortex of mice of the indicated genotypes. (E) Plaque size and plaque density (normalized to the control) in the cerebral cortex of the indicated genotypes. (F) Percentages of the areas occupied by plaques in the indicated cortical layers. (G) GFAP immunostaining in the cerebral cortex of mice of the indicated genotypes. Notice the decreased GFAP-positive labeling and reactivity of astrocytes in the cerebral cortex of 5xFAD mice expressing neuronal E2F4DN (arrows). (H) Percentages of the areas occupied by GFAP immunostaining in the indicated cortical layers. Numbers refer to the different cortical layers. Inserts show high magnifications of the indicated dashed boxes. *p<0.05; **p<0.01 (Student’s t test). Scale bar: 100 μm.
The predicted reduction of phagocytic capacity of microglia was consistent with the larger size of the Aβ deposits in the cerebral cortex of 5xFAD/E2F4DN mice, as compared with control 5xFAD/EGFP mice, without affecting their density (Fig. 3d,e). This effect, which was also observed in the hippocampus (Fig. S2c,d), was most evident in the cortical layer 5 (Fig. 3f). The increased plaque size was neither due to an increase of Aβ production (Fig. S3) nor anomalous distribution of microglial cells, which were found surrounding the Aβ deposits in the 5xFAD/E2F4DN condition (Fig. 3d). This finding confirmed our previous observation that Aβ deposits are enlarged in the cerebral cortex of 6 month-old 5xFAD/E2F4DN mice, as evidenced by Aβ-specific immunostaining (27).
Neuroinflammation in 5xFAD mice is accompanied by reactive astrogliosis (44) (see Fig. S4). We therefore performed GFAP immunostaining in cerebral tissue from 5xFAD/EGFP and 5x/E2F4DN mice of 3 months of age. This analysis indicated that neuronal expression of E2F4DN reduces the area of GFAP immunoreactitivity in the cerebral cortex (Fig. 3g) of 5xFAD mice. This reduction was statistically significant in cortical layer 6 (Fig. 3h), and also in the hippocampus (Fig. S2e,f). These results contrast with the increase of the Gfap transcript detected by RNA-seq (Table S1) and qPCR in both cerebral cortex (Fig. 2f) and hippocampus (Fig. S1c), suggesting that posttrancriptional mechanisms could regulate GFAP expression (45).
We conclude that neuronal E2F4DN expression favors a DAM-like phenotype with attenuated phagocytic and likely cytotoxic capacity as well as reduced astrogliosis, which altogether may benefit neuronal welfare.
Control by E2F4DN of gene networks involved in processing, accumulation and toxicity of Aβ
Among the genes whose expression is increased in the cerebral cortex of 5xFAD/E2F4DN mice, a small group is involved in preventing processing, accumulation and toxicity of Aβ (Fig. 4a). This group includes Grn, which encodes Progranulin, a neurotrophic growth factor that protects against Aβ deposition and toxicity (46); Hspb8, which encodes a heat shock protein that inhibits Aβ aggregation and toxicity (47); and St14, a microglial gene (32) that encodes Matriptase, a type II transmembrane serine protease that cleaves APP and reduces its processing to Aβ (48). We selected Hspb8 to confirm that the hippocampus shows a similar gene network (Fig. S1d). The upregulation in the cerebral cortex of 5xFAD/E2F4DN mice of other genes involved in Aβ aggregation and processing was also demonstrated by qPCR (Fig. 4b). They include Mme, which encodes neprilysin, an Aβ-degrading enzyme (49); A2m, which encodes α2-macroglobulin, an extracellular chaperone that inhibits amyloid formation (50); and Plaur, which encodes urokinase-type plasminogen activator. This protein can be a protective factor for degradation and clearance of Aβ (51). Adcyap1, which encodes pituitary adenylate cyclase activating polypeptide (PACAP), is also upregulated in the cerebral cortex of 5xFAD mice expressing neuronal E2F4DN (Fig. 4c). This neuropeptide, which is reduced in the brain of Alzheimer patients (52) and protects neurons against β-amyloid toxicity (52, 53), has been proposed as a therapy for AD (52). PACAP has been shown to stimulate the non-amyloidogenic processing of APP and to increase the expression of BDNF and of the antiapoptotic Bcl-2 protein (53). PACAP can also enhance the expression of the Aβ-degrading enzyme neprilysin in the mouse brain (53). Adcy7, which encodes the major adenylate cyclase isoform downstream of PACAP (54), is also increased in the cerebral cortex of 5xFAD/E2F4DN mice (Fig. 4c). Overall, the observed gene network favoring reduced processing, accumulation, and toxicity of Aβ likely facilitates brain welfare while accounting for size control of Aβ deposits in a genetic context supporting reduced phagocytic capacity of microglia.

They include genes preventing processing, accumulation and toxicity of Aβ (A), genes not detected by RNA-seq that are involved in Aβ aggregation and processing (B), genes involved in the PACAP signaling pathway (C), Thbs4, which regulates neurite outgrowth and synapse formation (D), Irs2, which regulates insulin signaling (E), genes involved in oxidative stress regulation (F), genes involved in vascular integrity (G), and brain welfare markers (H). Relative gene expression was normalized to Rps18 rRNA levels and expressed as 2ΔCt (obtained values were adjusted by the factor indicated between brackets). *p<0.05; **p<0.01; ***p<0.001 (Unbalanced two-way ANOVA, followed by post hoc post hoc Student’s t test).
Other pathological processes modulated by E2F4DN
Neuronal expression of E2F4DN also modulates groups of genes that participate in biological processes highly relevant for AD, including synaptic function, glucose metabolism, oxidative stress, and endothelial cell function. Selected genes from these groups were analyzed by qPCR.
Synaptic dystrophy could be modulated by E2F4DN in 5xFAD mice though the upregulation of Thbs4 (Fig. 4d), which encodes TSP-4, a member of the thrombospondin family that regulates neurite outgrowth and synapse formation, and has been related to AD (55). The upregulation of Thbs4 gene expression suggests that synaptic function is potentiated in 5xFAD mice expressing neuronal E2F4DN. Accordingly, we found that synaptophysin, a known marker used for synapse quantification (56), is upregulated in the hippocampus of 5xFAD/E2F4DN mice (Fig. S5). Irs2 encodes insulin receptor substrate 2, a mediator of the insulin signaling pathway, which is compromised in AD (57). We found that E2F4DN increases Irs2 expression in 5xFAD mice (Fig. 4e). Oxidative stress might be attenuated by neuronal E2F4DN since Nnt shows a tendency (p=0.054) to become upregulated in the cerebral cortex of 5xFAD/E2F4DN mice (Fig. 4f). This gene encodes nicotinamide nucleotide transhydrogenase, an integral protein of the inner mitochondrial membrane involved in antioxidant defense in this organelle (58). A hypothetical decrease of oxidative levels in the cerebral cortex of 5xFAD/E2F4DN mice is consistent with the downregulation of Aldh1a1 expression in this tissue (Fig. 4f). This gene encodes an aldehyde dehydrogenase that becomes upregulated under oxidative stress conditions (59). Another evidence for the pathological improvement induced by neuronal E2F4DN is the upregulation of Klf4 (Fig. 4g), which encodes a protein that potentiates endothelial and vascular integrity (60). This upregulation could counteract chronic hypoperfusion known to occur in AD, which is associated to morphological alteration and elimination of blood vessels (9). Egfl6, which is decreased in 5xFAD control mice and becomes upregulated by E2F4DN, encodes an extracellular matrix protein involved in angiogenesis (61). This could counteract the blood flow reduction observed in AD. The beneficial effects of neuronal E2F4DN on brain welfare were confirmed by the reduction in the cerebral cortex of 5xFAD/E2F4DN mice of metabolic stress response markers including Hddc3, a putative marker of cell starvation (62), and Stbd1, a glycophagy marker (63) (Fig. 4h). We found that a selected subset of genes described above show a similar modulation by E2F4DN in the hippocampus (Fig. S1e-f).
Among other genes whose expression is modulated by E2F4DN with unclear purpose are Adi1, Barx2, Cfap46, and Cfap54. Adi1, which is upregulated in the cerebral cortex of 5xFAD/E2F4DN mice (Fig. S6a), encodes acireductone dioxygenase 1, an enzyme that participates in the metabolism of methionine and is downregulated in subjects with AD (64). Barx2 is downregulated in the cerebral cortex of 5xFAD/E2F4DN mice (Fig. S6a). The interaction of BARX2 with members of the CREB family (65), the latter known to participate in long-term potentiation and memory consolidation (66), makes us to speculate that this molecule could have a relevant role in AD. Finally, Cfap46 and Cfap54, which encode two structural proteins from motile cilia (67), have opposed modulation by E2F4DN (Fig. S6b). In the brain, motile cilia enable cerebrospinal fluid (CSF) flow in the ventricular system (68), and CSF circulatory dysfunction has been postulated as a relevant etiological factor for AD since this would result in Aβ and phospho-tau accumulation (69). Whether or not E2F4DN-dependent enrichment of CFAP54 with respect to CFAP46 may play a beneficial role for CSF circulation is an appealing hypothesis to be considered.
E2F4DN expression prevents neuronal tetraploidization in the cerebral cortex of 5xFAD mice
Neuronal tetraploidy could be an important etiological factor in AD (70). To study whether neuronal expression of E2F4DN can prevent AD-associated NT, we analyzed this parameter in descendants of crosses between 5xFAD mice and either E2F4DN or EGFP mice at 3 months of age. The proportion of tetraploid nuclei for each genotype was normalized to the value obtained in the cerebral cortex wild-type (WT) mice of 2 months of age (71), which were used as an internal control in all the experiments (70). These analyses indicated that NT was increased in 5xFAD/EGFP mice when compared with WT/EGFP littermates (Fig. 5a), in agreement with previous observations in APP/PS1 mice (70). This effect was blocked in the presence of E2F4DN (Fig. 5a). We therefore conclude that E2F4DN is able to prevent NT, as expected from its capacity to prevent cell cycle re-entry in differentiating chick retinal neurons (25).

(A) NT quantification, normalized to NT levels in the cerebral cortex of 2-month-old WT mice, in cell nuclear extracts from cerebral cortex of 3-month-old littermates from crosses between 5xFAD transgenic mice and either homozygous EGFP mice or E2F4DN mice. (B) Body weight in one-year-old male littermates from crosses between 5xFAD transgenic mice and either homozygous EGFP mice or homozygous E2F4DN mice. (C) Left: Percentage of alternation observed in 5-month-old littermates from crosses between 5xFAD transgenic mice and either homozygous EGFP mice or homozygous E2F4DN mice. Random alternation (50%) indicates full memory loss. E2F4DN expression prevents spatial working memory impairment in 5xFAD mice as evaluated by the spontaneous alternation Y-maze test. Right: Number of arms entered by 5 month-old littermates from crosses between 5xFAD transgenic mice and either homozygous EGFP mice or homozygous E2F4DN mice. *p<0.05; ***p<0.001 (Unbalanced two-way ANOVA, followed by post hoc Student’s t test).
E2F4DN expression prevents body weight loss in 5xFAD mice
A previous report demonstrated that 5xFAD mice show progressive body weight loss, starting at 9 months of age (72), a pathological effect also observed in AD patients (1). In accordance with (72), we found a loss of body weight when 5xFAD/EGFP mice were compared with WT/EGFP mice at 1 year of age (Fig. 5b). In contrast, no body weight loss was detected in the presence of E2F4DN (Fig. 5b). This indicates that the expression of E2F4DN can reverse this somatic phenotype in 5xFAD mice.
E2F4DN expression prevents spatial memory deficits in 5xFAD mice
5xFAD mice of 4-5 months of age display cognitive impairment, evidenced by the spontaneous alternation Y-maze paradigm (28). We therefore tested whether E2F4DN expression reverses this phenotype. To this aim, spontaneous alternation performance in the Y-maze test was analyzed in 5-month-old descendants from both 5xFAD/EGFP and 5xFAD/E2F4DN crosses. This analysis confirmed that 5xFAD mice show reduced probability of alternation under control conditions (Fig. 5c, left). In contrast, the expression of E2F4DN in neurons fully reversed the spatial memory deficits of 5xFAD mice (Fig. 5c, left). These results were independent of the number of arms that were entered in either WT/EGFP vs.
5xFAD/EGFP or WT/E2F4DN vs. 5xFAD/E2F4DN mice (Fig. 5c, right). Therefore, working memory is improved in 5xFAD mice with neuronal expression of E2F4DN. This effect does not rely on better locomotor activity, as evidenced by the activity cage test (Fig. S7a,b), or motor coordination, as measured by a rotarod test (Fig. S7c).
Discussion
Although E2F4 has been recognized for decades as a transcription factor with a crucial role in the control of cell quiescence, recent evidence indicates that it can also fulfil multiple homeostatic functions (73). In Aβ-stressed neurons, E2F4 could play a protective role (74) due to its potential capacity to bind the regulatory domains of over 7,000 genes, and to modulate several transcriptional networks (20).
Our results support this hypothesis since the mutation of the conserved Thr249/T251 motif of mouse E2F4, which is susceptible to be phosphorylated (21), is crucial for maintaining neuronal quiescence and brain homeostasis in the presence of elevated levels of Aβ. This is consistent with our previous observation that E2F4 is associated with phosphoThr immunoreactivity in cortical neurons from APP/PS1 mice (27), and that E2F4 phosphorylation can lead to cell cycle progression (25, 75). In stressed neurons, E2F4 phosphorylation could favor the expression of mitotic regulators (25, 74), and cell cycle-independent genes involved in the etiology of AD, thus explaining the capacity of E2F4DN to restore normal E2F4 function and prevent several AD-associated processes. Although the conserved Thr249/Thr251 motif can be phosphorylated by p38MAPK (25), a stress kinase upregulated in AD (26), we cannot rule out that other related stress kinases could also lead to E2F4 phosphorylation in AD.
Microglial cells function as a sensor of changes in their environment and respond to such changes providing neuroprotection, but an exacerbation of this essential function leads to neurodegeneration. Correcting this imbalance may be a potential mode for therapy (76). We have shown that neuronal expression of E2F4DN modulates both glial cell morphology and expression of genes involved in microglial function, likely through well-established mechanisms of bidirectional neuron-glia communication (77). The resulting microglial phenotype might favor brain homeostasis reducing adverse effects derived from cytotoxicity and Aβ phagocytosis (4, 77).
The observation that Apoe expression remains at low levels in the cerebral cortex of 5xFAD/E2F4DN mice while other DAM-associated genes become upregulated is consistent with a putative reduced phagocytic capacity of microglia. This is relevant for Aβ processing since ApoE plays a critical role in the ability of microglia to take up and degrade Aβ (78). Reduced Aβ phagocytic capacity likely reduces NLRP3 inflammasome activation in microglia and the downstream effects on Aβ and tau pathology (5). Although the reduced clearance of fibrillar Aβ reduces the neuroinflammatory response, it results in Aβ accumulation, which is likely attenuated in 5xFAD/E2F4DN mice by the expression of a repertoire of genes that prevent its processing, accumulation and toxicity. Nevertheless, cognition is compatible with the presence of extensive Aβ deposition, as evidence from individuals with asymptomatic AD (29), and current view stablishes that Aβ is necessary but not sufficient for the etiology of AD, in which microglia plays a prominent role (4). As in our study, (79) have shown strong Aβ accumulation in a clinical case with resistance to familial AD in correlation with an APOE3 mutation in homozygosis, which led the authors to propose that reduced ApoE activity is likely to prevent cognitive deficits.
Other pathological mechanisms could also be attenuated by E2F4DN in 5xFAD mice, as relevant genes involved in the complex etiology of AD were found to be positively modulated. This includes regulators of synaptic function, glucose metabolism, oxidative stress, and endothelial cell function. Of relevance is also the demonstration that NT is prevented by neuronal E2F4DN expression. Cell cycle reentry, together with neuroinflammation, has been proposed to be major drivers of AD (10), likely due to its capacity to induce NFTs, extracellular deposits of Aβ, gliosis, synaptic dysfunction and delayed neuronal cell death [reviewed by (12)]. Furthermore, NT triggers synaptic dysfunction (80) and may affect neuronal structure and function (12). Our results are consistent with the observation that prevention of NT in the cerebral cortex of aged E2f1-/- mice correlates with enhanced cognition (70).
E2F4DN expression in neurons was able to reverse the weight loss phenotype observed in 5xFAD mice (72). Weight loss is a common symptom of AD (1), likely associated to metabolic alterations (81). This latter view is consistent with the increase of the resting metabolic rate observed in a mouse model of tau deposition (82), as well as the early metabolic deficits detected in transgenic mice overexpressing APP, in association with hypothalamic dysfunction (83). At present, it is uncertain whether the effect of E2F4DN on AD-associated metabolic alterations is directly due to a hypothetical capacity to block NT in neurons involved in sensing leptin, an adipocytokine that regulates energy metabolism and appetite (84). Alternatively, E2F4DN might act as a transcription factor on metabolism regulating pathways. Indeed, E2F4 is known to be regulated by insulin signaling in preadipocytes (85). Therefore, E2F4 seems to be linked to multiple pathways involved in obesity and energy metabolism, and this property may underlie its capacity to reverse weight loss in AD.
In this study, we have demonstrated that E2F4DN expression prevents spatial learning deficits observed in 5xFAD mice, evaluated with the spontaneous alternation Y-maze test (28). Since Aβ deposition was not prevented by neuronal expression of E2F4DN, cognitive recovery in 5xFAD mice cannot result from Aβ accumulation blockage. The latter has been claimed as the reason why therapeutic approaches aimed at reducing amyloid burden that work in transgenic mice are not effective when translated to AD patients (86).
In contrast, our results suggest that E2F4DN is useful as a multifactorial therapeutic agent for AD. When E2F4DN is expressed in neurons, multiple neuropathological and somatic alterations observed in 5xFAD mice become attenuated without triggering major side effects. The absence of side effects was expected, as E2F4 is already expressed in neurons from 5xFAD mice. A major challenge for E2F4DN as a therapeutic agent is the way for neuronal delivery of the molecule in vivo, both in AD mouse models and AD patients. We propose E2F4DN-based gene therapy as a realistic approach. Gene therapy is usually regarded as a process whereby WT genes are delivered in a tissue to replace abnormal genes that cause pathological effects. In our case, functional recovery would be obtained by expressing an E2F4 form unable to become Thr phosphorylated, thus counteracting the pathological environment that favors E2F4 phosphorylation. In this way, neuronal E2F4DN expression could efficiently target the complex etiology of AD (15-18), thus becoming a promising molecule for a successful therapy against this devastating disease.
Materials and Methods
Experimental design
Our study aimed to investigate the effect of neuronal expression of E2F4DN on the neuropathological phenotype observed in 5xFAD mice. To this end, we generated a novel knock-in mouse strain expressing myc-tagged E2F4DN under the control of the neuron-specific Mapt gene (30). The expression of E2F4DN-myc was evaluated by western blot in cortical extracts from E2F4DN mice, as compared with WT cortical extracts. The expression of the E2F4DN-myc transgene in neurons was then evaluated in cryosections from WT and E2F4DN mouse brain, immunostained with an anti-myc antibody. As a control for the rest of studies, we used Mapt:EGFP knock-in mice (30). Mice from both knock-in mouse strains were crossed with 5xFAD mice and their progenies were randomly used in seven different studies. Firstly, we performed an RNA-seq analysis using total RNA from the cerebral cortex of two 5xFAD/EGFP and two 5xFAD/E2F4DN mice to identify differentially expressed genes. The set of differentially-expressed genes found in this study, which were validated by qPCR (see below), were compared to the differentially-expressed genes described by (32) in the cerebral cortex of APP/PS2 mice, an equivalent mouse model of AD. Then, two sets of bioinformatics analyses were performed. On the one hand, we explored the function of the differentially-expressed genes in APP/PS2 mice that are either common or non-common to those modulated by E2F4DN in 5xFAD mice. For this purpose, we used the DAVID bioinformatics platform. This analysis provided important information about the pathological processes modulated by E2F4DN. On the other hand, by using the Enrichr bioinformatics platform we browsed the MGI Mammalian Phenotype database to identify major phenotypes associated with the protein-encoding genes that are modulated by E2F4DN in the cerebral cortex of 5xFAD mice, but are absent in the study by (32). This analysis was used to confirm the conclusions obtained in the former bioinformatics study. Secondly, we studied by qPCR the expression patterns of an ample set of genes differentially expressed in the cerebral cortex of 5xFAD mice expressing E2F4DN. These genes were chosen because of their relationship with pathways known to participate in AD. For this analysis, we synthesized cDNAs from the cerebral cortex and hippocampus of WT/EGFP, WT/E2F4DN, 5xFAD/EGFP, and 5xFAD/E2F4DN mice (at least 4 mice per group). These cDNAs were subjected to qPCR, and the statistical significance of the results was tested using unbalanced ANOVA followed by post hoc Student’s t tests. This analysis confirmed the E2F4DN-dependent modulation observed by RNA-seq. Thirdly, we performed Iba1-specific and GFAP-specific immunohistochemistry in brain sections from 5xFAD/EGFP, and 5xFAD/E2F4DN mice to characterize morphological alterations in glial cells that could confirm the molecular changes observed by qPCR. For this analysis, we employed at least four mice per group. These sections were also used for quantification of Thioflavin S-labeled Aβ deposits. Fourthly, we performed western blots with hippocampal extracts from 5xFAD/EGFP and 5xFAD/E2F4DN mice (n=3 per group) to characterize the levels of expression of both APP and soluble forms of Aβ. This same technique was performed with hippocampal extracts from WT/EGFP, WT/E2F4DN, 5xFAD/EGFP, and 5xFAD/E2F4DN mice (n=3 per group) to characterize the levels of expression of synaptophysin, a well-known synaptic marker (56). Fifthly, we analyzed the proportion of tetraploid neurons in nuclear extracts immunostained with the neuronal marker NeuN from WT/EGFP, WT/E2F4DN, 5xFAD/EGFP, and 5xFAD/E2F4DN mice (at least twelve mice per group, to obtain robust statistics). This analysis was performed using a well-characterized flow cytometry-based procedure developed in our laboratory (70, 71). Sixthly, we weighted a cohort of WT/EGFP, WT/E2F4DN, 5xFAD/EGFP, and 5xFAD/E2F4DN mice at one year of age (at least 10 mice per group, to obtain robust statistics). Finally, we evaluated the cognitive capacity of WT/EGFP, WT/E2F4DN, 5xFAD/EGFP, and 5xFAD/E2F4DN mice at five months of age (at least eleven mice per group, to obtain robust statistics) by performing the spontaneous alternation Y-maze test.
Mice
Experimental procedures with mice were approved by the CSIC animal ethics committee and the Autonomous Government of Madrid, in compliance with the Spanish and European Union guidelines. Double transgenic mice in C57BL/6J genetic background expressing under the control of the Thy1 promoter both mutant human APP695 with the Swedish (K670N, M671L), Florida (I716V), and London (V717I) FAD mutations and human PS1 harboring the M146L and L286V FAD mutations (Tg6799 or 5xFAD mice) (28) were purchased from The Jackson Laboratory (strain #008730). 5xFAD mice were genotyped as indicated by The Jackson Laboratory. A 5xFAD mouse strain was obtained after repeated inbreeding of originally hemizygous mice. Homozygous Mapttm1(EGFP)Klt knock-in mice expressing enhanced green fluorescent protein (EGFP) in neurons (EGFP mice) (30), were purchased from The Jackson Laboratory (strain #004779). EGFP mice have a target mutation in the Mapt gene, in which the coding sequence of EGFP has been inserted into the first exon, thus disrupting the expression of the Tau protein. This results in the neuron-specific expression of cytoplasmic EGFP. Tau is expressed at high levels in neurons (87), and homozygous mice mutant for tau are viable, fertile and display no gross morphological abnormalities in the central or peripheral nervous systems (30). Homozygous EGFP mice are viable, fertile, normal in size and do not display any gross physical or behavioral abnormalities. EGFP mice were genotyped as indicated by The Jackson Laboratory. These mice were used in this study as a control for E2F4DN mice. Homozygous EGFP mice were bred with hemizygous 5xFAD mice to generate littermates constituted of hemizygous EGFP mice with or without the 5xFAD transgene. Mapttm(mE2F4DN-myc) knock-in mice (E2F4DN mice) were generated following the procedure described by (30). These mice express a dominant negative form of E2F4 equivalent to the mutant E2F4 used to prevent NT in chick neurons (25). To this aim, a cassette containing the coding sequence of mouse E2F4 with the Thr249Ala/Thr251Ala mutations followed by the c-Myc tag, the Pgk-1 polyadenylation signal, and the G418-selectable marker Pgk-Neor was inserted into the NcoI site of a plasmid containing exon 1 of the Mapt gene and 8.0 kb flanking genomic sequence. The linearized targeting vector was electroporated into 129Sv-derived R1 embryonic stem cells. One hundred eighty-two G418-resistant colonies were analyzed by genomic PCR using Taq DNA polymerase (BioTools) to verify the upstream inserted sequence using primers #1 and #2 (5’AGGAGGCAGAAACAAGTGGA3’ and 5’ACACGAACTTGGTGGTGAGA3’, respectively; amplicon: 2,160 bp) (Fig. 1A). Nine of the analyzed clones produced the expected band. These clones were further analyzed by genomic PCR using Long Amp Taq DNA polymerase (New England Biolabs) to check the downstream inserted sequence. For this analysis the following primers were used: 5’GGCGCCCGGTTCTTTTTGTC3’ (primer #3) and 5’CACACAGCTAGTCCACAAAG3’ (primer #4) (amplicon 7,951 bp) (Fig. 1A). Two clones (#138 and #174) were found to produce the expected band. After confirming the euploidy of this latter clone, it was used to make chimeras by injection into C57BL/6 blastocysts, and four male high-percentage chimeras were mated to C57BL/6 females. Genomic PCR using the oligonucleotides that amplify the upstream sequence (primers #1 and #2) indicated that agouti offspring of chimeric males (n=25) carried the transgene at the expected Mendelian frequency. Subsequent progeny was analyzed by genomic PCR with primers #1 and #2 (E2F4DN band) and the WT primers described by The Jackson Laboratory for EGFP mice (5’-CTCAGCATCCCACCTGTAAC-3’ and 5’-CCAGTTGTGTATGTCCACCC-3’). The knock-in strain was maintained on a mixed background of C57BL/6 and 129Sv or backcrossed to the C57BL/6 background. Homozygous E2F4DN mice were created by inbreeding mice containing one copy of the E2F4DN transgene. Homozygous E2F4DN mice are viable, fertile, normal in size, and do not display any gross physical or behavioral abnormalities, even though the tau protein has been deleted (30). Homozygous E2F4DN mice were bred with hemizygous 5xFAD mice to generate littermates constituted of hemizygous E2F4DN mice with or without the 5xFAD transgene. Analyses were performed in hemizygous mice for both Egfp and E2f4dn transgenes to avoid the observed effects of full Mapt null mutation in the phenotype of APP and APP/PS1 transgenic mice (88). E2F4DN mice are available upon request for research purposes other than neurological and neurodegenerative diseases.
Antibodies
Rabbit anti-Myc tag polyclonal antibody (pAb) (ab9106; Abcam) at 1/1,000 for western blot and at 1/500 for immunohistochemistry. Mouse anti-α-tubulin monoclonal antibody (mAb) clone DM1A (ab7291; Abcam) was used at 1/10,000 for western blot. The rabbit anti-NeuN pAb (ABN78, Merck Millipore) was diluted 1/800 for flow cytometry and 1/1,000 for immunohistochemistry. The mouse anti-NeuN mAb, clone A60 (MAB377; Merck Millipore) was used at 1/1,600 dilution for immunohistochemistry. The rabbit anti-GFAP pAb (ab7260, Abcam) was diluted 1/1,000 for immunohistochemistry. The rabbit anti-Iba1 pAb (019-19741, Wako) was used at 1/800 dilution. The mouse anti-Synaptophysin mAb SY38 (PROGEN) was diluted 1:500 for western blot. The rabbit anti-Lamin B1 mAb [EPR8985(B)] (abcam) was used at a 1:5,000 dilution for western blot. The rabbit anti-β-Amyloid pAb #2454 (Cell Signaling Technology) was diluted 1:1,000 for western blot. The APP mAb 22C11 (Invitrogen) was used at a dilution of 1:3,000 for western blot.
The donkey anti-rabbit IgG (H+L) highly cross-adsorbed secondary antibody, Alexa Fluor 488 (Invitrogen) was used at 1/1,000 dilution for immunohistochemistry and 1/400 for flow cytometry. The goat anti-mouse IgG (H+L) cross-adsorbed secondary antibody, Alexa Fluor 568 (Invitrogen) was diluted 1/1,000 for inmunohistochemistry. The IRDye 800CW Goat anti-Rabbit IgG (H + L) and IRDye 680RD Goat anti-Mouse IgG (H + L) antibodies (LI-COR) were diluted 1/15,000 for western blot.
RNA extraction and cDNA synthesis
Total RNA was extracted using QIAzol Reagent (Qiagen), and cDNA was synthesized using SuperScript IV Reverse Transcriptase (ThermoFisher Scientific) following the indications of the manufacturer.
RNA extraction and RNA-Seq library preparation
Total RNA was extracted from the cerebral cortex of two 5xFAD/EGFP mice and two 5xFAD/E2F4DN mice using TriZOL (ThermoFisher Scientific) according to the recommended protocol. Residual genomic DNA was removed with DNase I recombinant, RNase-free (Roche), following the manufacturer’s instructions. Concentration and quality of the total RNAs were measured using an Agilent Bioanalyzer 2100 using RNA 6000 nano Chips (Agilent Technologies). All samples had an RNA integrity value of 7 or greater. 1000 ng of RNA from the combination of two mice of each genotype were used for each RNA-Seq library, which was created using the “NEBNext Ultra Directional RNA Library preparation kit for Illumina” (New England Biolabs) following the manufacturer’s instructions. We followed the indications of “Chapter 1: Protocol for use with NEBNext Poly(A) mRNA Magnetic Isolation Module”. We performed the library amplification included in the manufacturer’s instructions using a PCR of 12 cycles. RNA-Seq libraries quality was assessed using an Agilent Bioanalyzer and Agilent DNA7500 DNA chips to confirm that the insert sizes were 200–400 bp (average size: 299-317) for all the individual libraries. The obtained individual libraries were also quantified by an Agilent 2100 Bioanalyzer using a DNA7500 LabChip kit and an equimolecular pool of libraries were titrated by quantitative PCR using the “Kapa-SYBR FAST qPCR kit for LightCycler 480” (Kapa BioSystems) and a reference standard for quantification. The pool of libraries was denatured prior to be seeded on a flowcell at a density of 2.2 pM, where clusters were formed and sequenced using a “NextSeq™ 500 High Output Kit” (Illumina Inc.).
RNA sequencing and bioinformatic analysis of RNA-Seq data
RNA-Seq libraries were single-end sequenced in a 1×75 format using an Illumina NextSeq500 sequencer at the Genomic Unit of the Scientific Park of Madrid, Spain. The raw reads (FD: 34,860,735 reads, FT: 29,591,070 reads) passed the quality analysis performed with the tool FastQC. Trimmed reads were subsequently mapped to the Genome Reference Consortium Mouse Buildt 38 patch release 5 (GRCm38.p5) adding the EGFP sequence with the tool Bowtie (89), integrated in the Tophat suite (90). The resulting alignment files were used to generate a transcriptome assembly for each condition with the tool Cufflinks, and the expression levels were then calculated with the tool Cuffdiff together with the statistical significance of each observed change in expression (90). p<0.05 was used as a criterion for differential expression since FAD/EGFP vs FAD/E2F4DN comparison yielded to no gene tagged as significantly expressed by Cufflinks. As described in the main text, differential expression in several genes was confirmed by qPCR.
qPCR
Real-time RT-PCR was performed with the 7500 Real-Time PCR equipment (Applied Biosystems), using specific primers from PrimePCR SYBR Green Assay (BioRad) and the house keeping gene Rps18 (qMmuCED0045430 PrimePCR SYBR Green Assay; BioRad). ΔCt for treatment and control was calculated and then statistical significance was evaluated by post hoc Student’s t test in genes where two-way ANOVA analysis was found to be significant.
Tissue processing
After anesthetizing the mice with intraperitoneal sodium pentobarbital (Dolethal; Vetoquinol), administered at 50 mg/kg (body weight), they were transcardially perfused with PBS, and then with 4% paraformaldehyde (PFA). Brains were finally postfixed overnight at 4 °C with 4% PFA and cryoprotected by sinking in 30% sucrose in PBS at 4°C. Then, brains were embedded in either Tissue-Tek (Sakura), followed by freezing in dry ice to get cryosections (12-15 μm), or 3% agarose gels prepared in 0.1 phosphate buffer, pH 7.37, before cutting them with a vibratome (50 μm).
Thioflavin S staining
Vibratome sections were washed three times with phosphate-buffered saline (PBS) containing 0.4% Triton X-100 (Sigma-Aldrich) (0.4% PBTx), and then incubated for 30 min in the dark with 0.05% Thioflavin S (Sigma-Aldrich) in 50% ethanol (Merck). Finally, sections were washed twice with 50% ethanol, and once with distilled water. Then, sections were subjected to immunohistochemistry as described below.
Immunohistochemistry
Cryosections were permeabilized and blocked for 1 h at RT in 0.1% PBTx and 10% fetal calf serum (FCS; Invitrogen), and incubated O/N at 4 °C with the primary antibodies in 0.1% PBTx plus 1% FCS. After washing with 0.1% PBTx, the sections were incubated for 1 h at RT in 0.1% PBTx plus 1% FCS with the secondary antibodies. The sections were washed in 0.1% PBTx, and then they were incubated with 100 ng/ml 4′,6-Diamidine-2′-phenylindole dihydrochloride (DAPI; Sigma-Aldrich) in PBS before mounting with ImmunoSelect antifading mounting medium (Dianova). Vibratome sections were permeabilized and blocked in 0.4% PBTx containing 10% FCS for 3 h. Then, they were incubated overnight at 4 °C with the primary antibodies in 0.1% PBTx containing 1% FCS. After five washes of 20 min with 0.1% PBTx, sections were incubated with the secondary antibodies plus 100 ng/ml DAPI in 0.1% PBTx for 3 h at room temperature (RT). Sections were then washed five times with 0.1% PBTx, and mounted with ImmunoSelect antifading mounting medium.
Quenching of lipofuscin autofluorescence signal
Lipofuscin present in the brain of 6 month-old mice was quenched with TrueBlackTM Lipofuscin Autofluorescence Quencher (Biotium). Briefly, vibratome sections were washed once with PBS and treated for 30 s with TrueBlack 1x prepared in 70% ethanol. Finally, sections were washed three times with PBS, and then immunostained as described above.
Confocal microscopy and image analysis
Confocal images were acquired at 20x magnification with a Leica SP5 confocal microscope. Image analysis was performed using ImageJ (Fiji). Images used for the analysis (at least two mosaic images per tissue and animal) were maximum intensity projections, created as output images whose pixels correspond to the maximum value of each pixel position (in xy) across all stack images (z). For the analysis of the area occupied by GFAP and Iba1, threshold was determined to highlight the area to be quantified. If necessary, the region of interest (ROI) tool was then used to delimitate the different cortical layers. Quantification of soma size and density of Iba-1 labelled microglia was performed as described by (43). Briefly, image analysis was performed using Fiji with MorphoLibJ (v1.4.4) integrated library and plugin, applying a grey scale attribute opening filter (area minimum: 25 pixels; connectivity: 8) to an 8-bit average intensity projection. An opening morphological filter (1-pixel radius octagon) was then used effectively to separate microglia soma from processes, before a maximum entropy threshold was used to segment microglia soma from image background. Quantification of microglia soma was completed using the Fiji Analyse Particles function with a particle size threshold of 10 pixels, to exclude small pixel noise and extract information regarding microglial soma area. Microglia density was determined by dividing the total microglia population (258-342 cell somas per animal) by total analyzed area.
Western blot
Cortical and hippocampal extracts were obtained in cold extraction buffer [20 mM Tris-HCl pH 6.8, 10 mM β-mercaptoehanol (Sigma-Aldrich), 1 mM EDTA (Merck), 1% Triton X-100, 1% SDS (Sigma-Aldrich)] including 1x cOmplete Mini, EDTA-free, protease inhibitor cocktail (Roche) (one hemicortex in 500 μl extraction buffer). Extracts were centrifuged for 10 min at 14,000 xg (at 4 °C) and supernatants were then boiled for 5 min in Laemli buffer. Extracts were fractionated by SDS PAGE on 10% acrylamide gels and transferred to Immobilon-FL membranes (Millipore). The membranes were incubated for 1 h with Odyssey Blocking Buffer TBS (LI-COR) (OBB), and then incubated ON at 4 °C with the appropriate antibody in OBB containing 0.1% Tween 20. For Aβ analysis, hippocampal extracts prepared as described above were boiled for 5 min in 50 mM Tris-HCl pH 8.0 containing 12% Glicerol (Merck), 4% SDS, 0,01% Coomasie Brilliant Blue G-250 (Sigma-Aldrich), and 2% β-mercaptoethanol. Extracts were fractionated on 16.5% Mini-PROTEAN Tris-Tricine gels (Bio-Rad) (30 V for the first hour and then 125 V) using 1x Tris/Tricine/SDS Running Buffer (Bio-Rad) (cathode) and 200 mM Tris-HCl pH 9.0 (anode), and then transferred to Pierce Low-Fluorescence PVDF Transfer Membranes (0.2 μm) (ThermoFisher Scientific). The membranes were incubated for 1 h with Intercept Blocking Buffer TBS (LI-COR) (IBB), and then incubated ON at 4 °C with the rabbit anti-β-Amyloid pAb (1/1000) in IBB containing 0.1% Tween 20. After washing the membranes five times in TBS containing 0.1% Tween 20 (TBS-T), they were incubated for 1 h at RT with a 1/15,000 dilution of secondary antibodies in OBB (IBB for Aβ analysis) containing 0.1% Tween 20. Finally, they were washed again with TBS-T as described above, and the protein bands were visualized using the Odyssey CLx Infrared Imaging System (LI-COR).
Cell nuclei isolation
Cell nuclei isolation was performed as described by (71). Briefly, fresh-frozen mouse cerebral hemicortices were placed in 2.5 ml ice-cold, DNase-free 0.1% PBTx and protease inhibitor cocktail (Roche) (nuclear isolation buffer). Cell nuclei were then isolated by mechanical disaggregation using a dounce homogenizer. Undissociated tissue was removed by centrifugation at 200xg for 1.5 min at 4°C. The supernatant was 8-fold diluted with nuclear isolation buffer and centrifuged at 400xg for 4 min at 4°C. Supernatant with cellular debris was discarded, and the pellet incubated at 4°C in 800-1,000 μl cold nuclear isolation buffer for at least 1 h, prior to mechanical disaggregation by gently swirl of the vial. The quality and purity of the isolated nuclei was analyzed microscopically after staining with 100 ng/ml DAPI.
Flow cytometry
Flow cytometry was carried out as described by (71). Immunostaining of cell nuclei was performed by adding both primary (rabbit anti-NeuN) and secondary (Alexa 448-coupled anti-Rabbit) to 400 μl of isolated unfixed nuclei containing 5% of fetal calf serum (FCS) and 1.25 mg/ml of BSA. In control samples, the primary antibody was excluded. Finally, the reaction was incubated O/N at 4°C in the dark. Immunostained nuclei (400 μl) were filtered through a 40-μm nylon filter, and the volume adjusted to 800-1000 μl with DNase-free 0.1% PBTx containing propidium iodide (PI; Sigma-Aldrich) and DNAse-free RNAse I (Sigma-Aldrich) at a final concentration of 40 μg/ml and 25 μg/ml, respectively, and incubated for 30 min at RT. The quality of the nuclei and specificity of immunostaining signal was checked by fluorescence microscopy. Flow cytometry was then carried out with a FACSAria I cytometer (BD Biosciences, San Diego, CA) equipped with a 488-nm Coherent Sapphire solid state and 633-nm JDS Uniphase HeNe air-cooled laser. Data were collected by using a linear digital signal process. The emission filters used were BP 530/30 for Alexa 488, and BP 616/23 for PI. Data were analyzed with FACSDiva (BD Biosciences) and Weasel 3.0.1 (Walter and Eliza Hall Institute of Medical Research) softwares, and displayed using biexponential scaling. Electronic compensation for fluorochrome spectral overlap during multi-color immunofluorescence analysis was carried out when needed. Cellular debris, which was clearly differentiated from nuclei due to its inability to incorporate PI, was gated and excluded from the analysis. DNA content histograms were generated excluding doublets and clumps by gating on the DNA pulse area versus its corresponding pulse height. The exclusion of doublets was confirmed by checking the DNA pulse area versus the pulse width of the selected population, and the percentage of tetraploid nuclei was quantified. A minimum of 15,000 and 20,000 nuclei were analyzed for the NeuN-positive population. The proportion of tetraploid nuclei was normalized to the value obtained in cell nuclei from control 2 month-old WT mice as shown by (70), which was used as an internal control in all the experiments.
Activity cage test
Exploratory locomotor activity was recorded using a VersaMax Animal Activity Monitoring System (AccuScan Instruments, Inc.) in an open field (40 cm × 40 cm) over a 10 min period. Infrared beams automatically record horizontal movements and rearing in the open field. The task analyzes the activity behavior by measuring the number of beams that are broken during the designated period of time. Ten trials repeated in two consecutive days (five trials/day) were performed for every animal and results were expressed as average number of broken beams per trial.
Rotarod test
Motor coordination was evaluated in a Rotarod apparatus (Ugo Basile) with increasing acceleration. The apparatus consisted of a horizontal motor-driven rotating rod in which the animals were placed perpendicular to the long axis of the rod, with the head directed against the direction of rotation so that the mouse has to progress forward to avoid falling. The trial was stopped when the animal fell down or after a maximum of 5 min. The time spent in the rotating rod was recorded for each animal and trial. Animals received a pretraining session to familiarize them with the procedure before evaluation. Thereafter, a total of six consecutive trials were done for every animal. Data are presented as the average time spent before falling from the apparatus.
Spontaneous alternation Y-maze test
Each mouse was placed into the center of a Y-maze apparatus (Panlab) and then allowed to freely explore the different arms during an 8 min session. The sequence of arms entered was recorded and working memory was measured as the percentage of alternation (p.a.), which was calculated as the number of triads containing entries in all three arms divided by all the triads and then multiplied by 100.
Bioinformatics analysis
The DAVID bioinformatics platform was used for Gene Ontology (GO) functional annotation of gene sets (GOTERM_BP_ALL). MGI Mammalian Phenotype (Level 4 2019) was browsed using the Enrichr bioinformatics platform (http://amp.pharm.mssm.edu/Enrichr).
Statistical analysis
Quantitative data are represented as the mean ± s.e.m. Statistical differences in experiments performed with transgenic mice on either EGFP or E2F4DN background were analyzed using two-tailed Student’s t test. Two-way ANOVA analysis was performed in qPCR-based experiments, followed by post hoc Student’s t test. Two-way Student’s t test was performed for the quantitative analysis of immune cells. Outliers, as evidenced by the Grubbs’ test (transgenic mice experiments) or Dixon’s Q test (qPCR), were eliminated from the analysis.
Funding
This work has been supported by Ministerio de Economía y Competitividad grant SAF2015-68488-R, Ministerio de Ciencia, Innovación y Universidades grant RTI2018-095030-B-I00, and a R&D contract between CSIC and Tetraneuron. N.L.S. holds a Torres Quevedo grant from Ministerio de Industria. M.R.L. holds an Industrial Doctorate grant from Ministerio de Economía, Industria y Competitividad.
Author contributions
N.L.S. designed and performed experiments and analyzed data;
M.R.L. performed experiments and analyzed data; C.T. performed experiments; A.G.G. performed experiments; J.M.F. designed the study, analyzed data, and wrote the paper.
Competing interests
J.M. Frade is shareholder (8.96% equity ownership) of Tetraneuron, a biotech company exploiting his patent on the blockade of NT by E2F4DN as a therapeutic approach against AD. N.L.S. received her salary from a R&D contract with Tetraneuron, and currently she works for this biotech company. M.R.L. and A.G.G. works for Tetraneuron.
Data and materials availability
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Availability of the E2F4DN mouse strain is subjected to material transfer agreement. Additional data related to this paper may be requested from the authors.
Supplementary Materials

They include DAM-specific genes (A), innate immune response genes (B), Gfap, an astrocyte marker (C), Hspb8, which encodes a heat shock protein that inhibits Aβ aggregation and toxicity (D), Klf4, a gene involved in vascular integrity (E), and brain welfare markers (F). Relative gene expression was normalized to Rps18 rRNA levels and expressed as 2ΔCt (obtained values were adjusted by the factor indicated between brackets). *p<0.05; **p<0.01; ***p<0.001 (unbalanced two-way ANOVA, followed by Student’s t test).

(A) Iba1 immunostaining in the hippocampus of mice of the indicated genotypes. (B) Percentage of the area occupied by Iba1 immunostaining in the hippocampus. (C) Thioflavin S/Iba1 co-labeling in the hippocampus of mice of the indicated genotypes. (D) Plaque density (normalized to the control) and plaque size in the hippocampus of the indicated genotypes. (E) GFAP immunostaining in the hippocampus of mice of the indicated genotypes. Notice the decreased GFAP-positive labeling of astrocytes in the hippocampus of 5xFAD mice expressing neuronal E2F4DN (arrows). (F) Percentage of the area occupied by GFAP immunostaining in the hippocampus. Inserts show high magnifications of the indicated dashed boxes. DAPI counterstaining is included to identify the hippocampus. *p<0.05; **p<0.01 (Student’s t test). Scale bar: 100 μm.
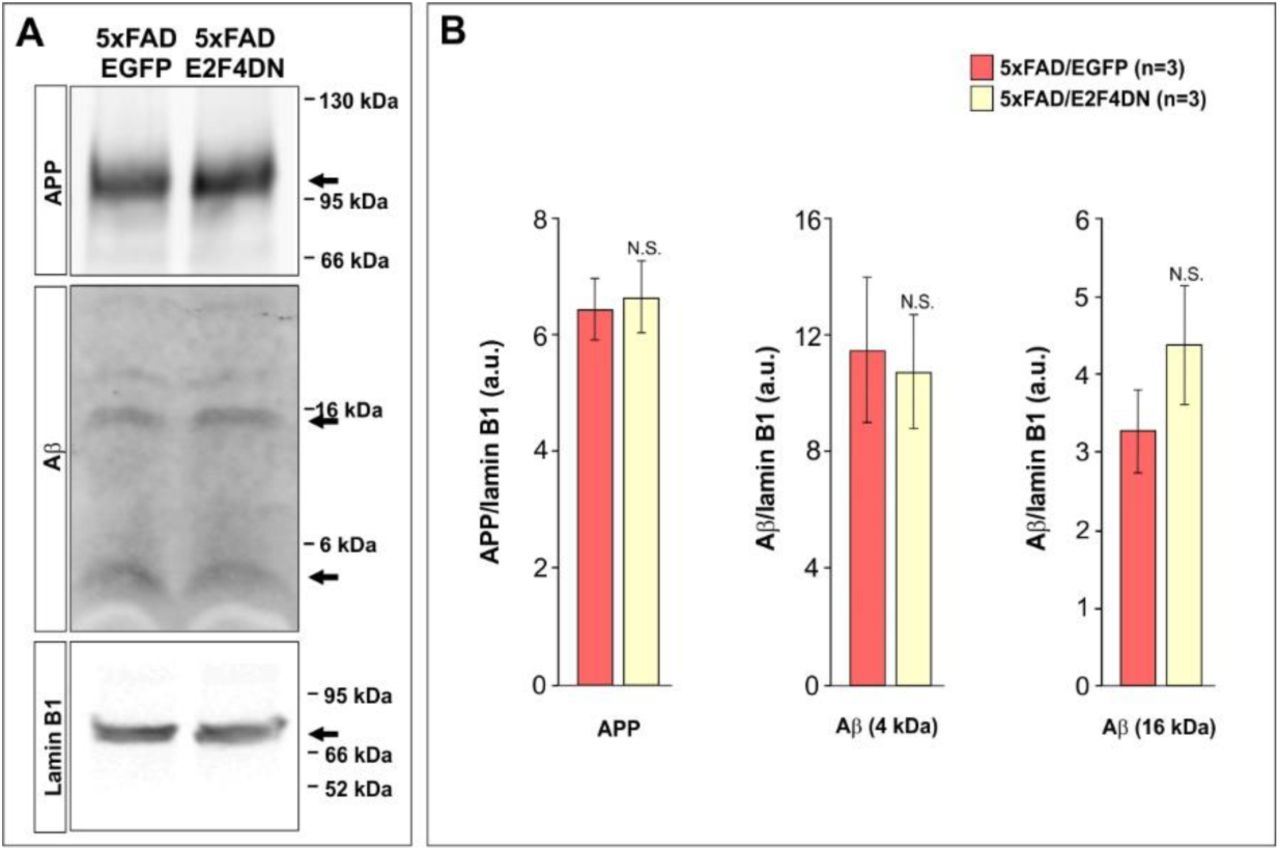
(A) Western blot analysis of hippocampal extracts from 5 month-old mice from the indicated genotypes using antibodies against APP, Aβ, and Lamin B1 (as a loading control). (B) Quantification of the western blots illustrated in A. Ratios against Lamin B1 are shown. N.S.: non-significant (Student’s t test).

Left panels: representative images of the expression pattern of GFAP at the indicated time points. Right panels illustrate the quantification of the percentage of area occupied by GFAP at the different temporal points. Inserts show high magnifications of the indicated dashed boxes. WT/EGFP: 2 months (n=3), 3 months (n=5), 6 months (n=4); 5xFAD/EGFP: 2 months (n=3), 3 months (n=5), 6 months (n=4); *p<0.05 (Student’s t test). Scale bar: 100 μm.

(A) Western blot analysis of hippocampal extracts from 3 month-old mice from the indicated genotypes using antibodies against Synaptophysin and Lamin B1 (as a loading control). (B) Quantification of the western blots illustrated in a. Ratios against Lamin B1 are shown. *p<0.05 (Student’s t test).

(A, B) Genes whose expression is modulated by E2F4DN in the hippocampus of 3 month-old mice of the indicated genotypes are shown. Relative gene expression was normalized to Rps18 rRNA levels and expressed as 2ΔCt (obtained values were adjusted by the factor indicated between brackets). *p<0.05; **p<0.01; ***p<0.001 (Unbalanced two-way ANOVA, followed by Student’s t test).

(A) Average number of infrared beams broken per trial over a 10 min period due to horizontal activity from crosses between heterozygous 5xFAD transgenic mice and either homozygous EGFP mice (left) or homozygous E2F4DN mice (right). (B) Average number of infrared beams broken per trial over a 10 min period due to vertical activity from crosses between heterozygous 5xFAD transgenic mice and either homozygous EGFP mice (left) or homozygous E2F4DN mice (right). (C) Motor coordination as measured by the rotarod text in 6-month-old littermates from crosses between heterozygous 5xFAD transgenic mice and either homozygous EGFP mice (left) or homozygous E2F4DN mice (right), measured as the time spent in the rotating rod before falling (latency). N.S.: non-significant (Unbalanced two-way ANOVA, followed by Student’s t test).
Table S1. Differentially expressed genes in the cerebral cortex of 5xFAD/E2F4DN and 5xFAD/EGFP mice. This includes the Egfp and E2f4-derived transgenes, 21 non-coding RNAs, 2 microRNAs, 2 transcripts encoding uncharacterized proteins, and 2 sets of related gene sequences encoding immunoglobulin heavy constant and T cell receptor α constant and joining moieties. A non-exhaustive list of references that associated the indicated genes with AD is included.
Table S2. Upregulated genes in the cerebral cortex of APP/PS2 transgenic mice, expressed in microglia, astrocytes, and neurons, as described by (32). Pink: Genes common between APP/PS2 and 5xFAD/E2F4DN. Blue: Genes in APP/PS2 but not in 5xFAD/E2F4DN.
Table S3. Bioinformatics analyses performed with differentially expressed genes in the cerebral cortex of 5xFAD/E2F4DN and 5xFAD/EGFP mice as compared with upregulated genes in the cerebral cortex of APP/PS2 transgenic mice. (Common Genes) Functional GO term annotation for biological processes (GOTERM_BP_ALL), from microglia-expressed genes common to 5xFAD/E2F4DN and APP/PS2 mice (32). (APP_PS2 Unique Genes) Functional GO term annotation for biological processes (GOTERM_BP_ALL) from microglia-specific genes unique in APP/PS2 mice (32). (MGI Mammalian Phenotype) MGI Mammalian Phenotype database browsed against the protein-encoding genes modulated by E2F4DN in the cerebral cortex of 5xFAD mice that are absent in the study by (32). Trac and Igha were included as representative for the sequences encoding immunoglobulin heavy constant and T cell receptor α constant and joining moieties shown in Table S1.
Acknowledgments
We thank Y.A. Barde for the plasmids to generate the E2F4DN knock-in mice, A. Arias, M.B. Pintado, M. García-Flores, V. Cano, and M.J. Román for their technical help, and C Sánchez-Puelles for her critical reading of the manuscript.
References and Notes
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.↵
- 89.↵
- 90.↵





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}