

ABSTRACT
Somatic mutations in cancer genomes are caused by multiple mutational processes each of which generates a characteristic mutational signature. Using 84,729,690 somatic mutations from 4,645 whole cancer genome and 19,184 exome sequences encompassing most cancer types we characterised 49 single base substitution, 11 doublet base substitution, four clustered base substitution, and 17 small insertion and deletion mutational signatures. The substantial dataset size compared to previous analyses enabled discovery of new signatures, separation of overlapping signatures and decomposition of signatures into components that may represent associated, but distinct, DNA damage, repair and/or replication mechanisms. Estimation of the contribution of each signature to the mutational catalogues of individual cancer genomes revealed associations with exogenous and endogenous exposures and defective DNA maintenance processes. However, many signatures are of unknown cause. This analysis provides a comprehensive perspective on the repertoire of mutational processes contributing to the development of human cancer.
INTRODUCTION
Somatic mutations in cancer genomes are caused by mutational processes of both exogenous and endogenous origins that have operated during the cell lineage between the fertilised egg and the cancer cell1. Each mutational process may involve components of DNA damage/modification, DNA repair and DNA replication, any of which may be normal or abnormal, and generates a characteristic mutational signature that may incorporate base substitutions, small insertions and deletions, genome rearrangements, and chromosome copy number changes2. The catalogue of mutations from an individual cancer genome may have been generated by multiple mutational processes and thus incorporate multiple superimposed mutational signatures. Therefore, in order to systematically characterise the mutational processes contributing to cancer, mathematical methods have been developed that can be used to (i) decipher mutational signatures from a set of somatic mutational catalogues, (ii) estimate the numbers of mutations attributable to each signature in each sample, and (iii) annotate each mutation class in each tumour with the probability of arising from each signature3–15.
Previous studies of multiple cancer types identified >30 single base substitution signatures, some of known but many of unknown aetiologies, some ubiquitous and others rare, some part of normal cell biology and others associated with abnormal exposures or operative during neoplastic progression7,16–27. Six genome rearrangement signatures have also been identified in breast cancer18 and further patterns of rearrangements have been described15,28–30. However, analysis of other mutation classes has been limited31,32.
Thus far, mutational signature analysis has predominantly used cancer exome sequences. However, the many fold greater numbers of somatic mutations in whole-genome sequences provide substantially increased power for decomposition, enabling better separation of partially correlated signatures and extraction of signatures which contribute relatively small numbers of mutations. Furthermore, technical artefacts and differences in sequencing technologies and mutation calling algorithms can themselves generate mutational signatures. Therefore, the uniformly processed and highly curated sets of all classes of somatic mutations from the 2,780 cancer genome sequences of the Pan Cancer Analysis of Whole-Genomes (PCAWG) project, combined with almost all other cancer genomes and exomes for which suitable mutational catalogues are publicly available, presents a notable opportunity to establish the repertoire of mutational signatures and to determine their activities across the range of cancer types.
RESULTS
Cancer genomes and somatic mutations
Somatic mutational catalogues from 23,829 samples of most cancer types, including the 2,780 highly curated PCAWG whole-genomes, 1,865 additional whole-genomes and 19,184 exomes were studied. From these, 79,793,266 somatic single base substitutions, 814,191 doublet base substitutions and 4,122,233 small insertions and deletions (indels) were analysed for mutational signatures, ~10-fold more mutations than any previous study (https://www.synapse.org/#!Synapse:syn11801889)4,16.
To enable mutational signature analysis classifications were developed for each type of mutation. For single base substitutions, the primary classification comprised 96 classes constituted by the six base substitutions C>A, C>G, C>T, T>A, T>C, T>G (in which the mutated base is represented by the pyrimidine of the Watson-Crick base pair) plus the flanking 5′ and 3′ bases. In some analyses, two flanking bases 5’ and 3’ to the mutated base were considered (generating 1,536 classes) or mutations within transcribed genome regions were selected and classified according to whether the pyrimidine of the mutated base pair fell on the transcribed or untranscribed strand (192 classes). A classification was also derived for doublet base substitutions (78 classes). Indels were classified as deletions or insertions and, when of a single base, as C or T and according to the length of the mononucleotide repeat tract in which they occurred. Longer indels were classified as occurring at repeats or with overlapping microhomology at deletion boundaries, and according to the size of indel, repeat, and microhomology (83 classes, https://www.synapse.org/#!Synapse:syn11726616)3,16.
Mutational signature analysis
The mutational catalogues from the 2,780 PCAWG whole-genome, 1,865 additional whole-genome, and 19,184 exome sequences of cancer were analysed separately. For each of these catalogue sets, signature extraction was conducted using methods based on nonnegative matrix factorisation (NMF)3,7 on each cancer type individually and also on all cancer types together. Analyses were carried out separately for single base substitutions (SBS signatures), doublet base substitutions (DBS signatures) and indels (ID signatures) and also for the three mutation types together (257 mutation classes, or 1697 if the 1536 SBS classification was employed) generating composite signatures.
Mutational signatures were extracted using two independently developed NMF-based methods: (i) SigProfiler, a further elaborated version of the framework used to generate the signatures shown in COSMIC3,16,18,33,34, and (ii) SignatureAnalyzer, based on a Bayesian variant of NMF used in several previous publications6,7,35,36. NMF determines both the signature profiles and the contributions of each signature to each cancer genome as part of its factorization of the input matrix of mutation spectra. However, given a substantial number of signatures and/or heterogeneous mutation burdens across samples, it is possible to reconstruct the mutations observed in a particular sample in multiple ways, often with very small and/or biologically implausible contributions from many signatures. Therefore, each method developed a separate procedure to estimate the contributions of signatures to each sample (Methods).
The results of the two methods exhibited many similarities. However, there were also noteworthy differences. The number of SBS signatures found in low mutation burden tumours in the PCAWG set (94.4% of cases that harbour 47% of mutations) was similar: 31 by SigProfiler and 35 by SignatureAnalyzer. The number of additional SBS signatures extracted from hyper-mutated PCAWG samples (5.6% of cases and 53% of mutations, https://www.synapse.org/#!Synapse:syn12016215), however, was different: 13 by SigProfiler and 25 by SignatureAnalyzer. There were also differences in SBS signature profiles, including among signatures found in low mutation burden cases. The latter primarily involved “flat”, relatively featureless signatures, which are mathematically challenging to deconvolute. Finally, there were differences in signature attributions to individual samples. In general, SignatureAnalyzer used more signatures to reconstruct the mutational profiles (Extended Data Figure 1, https://www.synapse.org/#!Synapse:syn12169204, https://www.synapse.org/#!Synapse:syn12177011) and the attribution to flat signatures was different, with SigProfiler assigning mutations to SBS5 and SBS40 and SignatureAnalyzer using combinations of multiple signatures (Extended Data Figure 2ab, https://www.synapse.org/#!Synapse:syn12169204). The DBS and ID signatures were generally similar between the two methods (Extended Data Figure 2cd). These comparisons provide a useful perspective on both the consistency and variability of signature extraction and attribution depending on the methodology used.
The final sets of reference mutational signatures were determined from the PCAWG analysis supplemented by additional signatures from the other datasets. Signatures were supported by the outcomes of analyses using the 192 and 1536 mutation classifications, the existence of individual cancer samples dominated by a particular signature, and, where available, prior experimental evidence for certain mutational signatures (Methods and https://www.synapse.org/#!Synapse:syn12009767). Each signature was allocated a number consistent with, and extending, the COSMIC annotation33. Some previous signatures split into multiple constituent signatures and these were numbered as before but with additional letter suffixes (eg, single SBS17 split into signatures SBS17a and SBS17b). DNA sequencing and analysis artefacts also generate mutational signatures, and we indicate which signatures are possible artefacts (https://www.synapse.org/#!Synapse:syn12009767) but do not present them below. However, future studies employing this signature set as a reference may consider utilizing artefact signatures for data quality control. The results of both SignatureAnalyzer and SigProfiler were used throughout the research reported here. However, for brevity and for continuity with the signature set previously displayed in COSMIC33, which has been widely used as a reference, SigProfiler results are outlined below and SignatureAnalyzer results are provided at (Extended Data Figures 3,4, https://www.synapse.org/#!Synapse:syn11738307).
Single base substitution (SBS) mutational signatures
There were substantial differences in numbers of SBSs between samples (ranging from hundreds to millions) and between cancer types, as previously observed16,37 (Figure 1). In total, 67 SBS mutational signatures were extracted, of which 49 were considered to be likely real (Figure 2, Methods, https://www.synapse.org/#!Synapse:syn12009783). Except for SBS25, all mutational signatures previously reported on COSMIC4,23,33 were confirmed in the new set of analyses (median cosine similarity between the newly derived signatures and those on COSMIC: 0.95, excluding “split” signatures which are discussed below; https://www.synapse.org/#!Synapse:syn12016215). SBS25 was previously found only in cell lines derived from Hodgkin lymphomas, some of which had been previously treated with chemotherapy, and, to our knowledge, no data from primary cancers of this type are currently available. The newly derived signatures showed much improved separation from each other and hence more distinct signature profiles, presumably due to the substantially increased statistical power of this analysis (https://www.synapse.org/#!Synapse:syn12009783).

Mutation burdens of single base substitutions, doublet base substitutions and small insertions and deletions for the 2,780 PCAWG tumors. Each sample is displayed according to its tumor type. Tumor types are ordered according to the median number of single base substitutions. The numbers of cases of each tumor type are shown. The proportions of each mutation subclass in each sample are shown as coloured bar charts.


Profiles of single base substitution, doublet base substitution and small insertion and deletion mutational signatures. The subclassifications of each mutation type (single base substitutions, 96 subtypes; doublet base substitutions, 78 subtypes; indels, 83 subtypes) are described in the main text. Magnified versions of signatures SBS4, DBS2 and ID3 (which are all associated with tobacco smoking) are shown to illustrate the positions of each mutation subtype on each plot.
Thirteen new likely real SBS signatures compared to the set previously described in COSMIC33 were extracted (excluding those that are the consequence of signature splitting). Some were in cancers with a previously unanalysed exogenous exposure (SBS42), some were in chemotherapy treated samples which have often been excluded from previous studies (SBS31, SBS32, SBS35) and some were rare and hence absent by chance from previous analyses (SBS36, SBS44). Others were more common, but contributed relatively few mutations to individual cancer genomes, or were similar to previously discovered signatures and thus not isolated from datasets based predominantly on cancer exome sequences (eg SBS38, SBS39, SBS40). Notably, SBS40 was extracted from kidney cancer in which it appears to be required for optimal reconstruction of mutational catalogues. It is a relatively featureless (“flat”) signature, with similarity to SBS5 and other flat signatures, and this may account for it only clearly emerging now with the availability of whole cancer genomes. SBS40 may contribute to other cancer types but its similarity to SBS5 renders this uncertain and larger datasets will be required to clarify the extent of its activity. For some new signatures there were plausible underlying aetiologies (Figure 3, Extended Data Figures 4,5): SBS31 and SBS35, prior platinum compound chemotherapy38; SBS32, prior azathioprine therapy; SBS36, inactivating germline or somatic mutations in MUTYH which encodes a component of the base excision repair machinery39,40; SBS38, additional effects of ultraviolet light (UV) exposure; SBS42, occupational exposure to haloalkanes27; SBS44, defective DNA mismatch repair due to MLH1 inactivation41. SBS33, SBS34, SBS37, SBS39, SBS40, and SBS41 are of unknown cause.
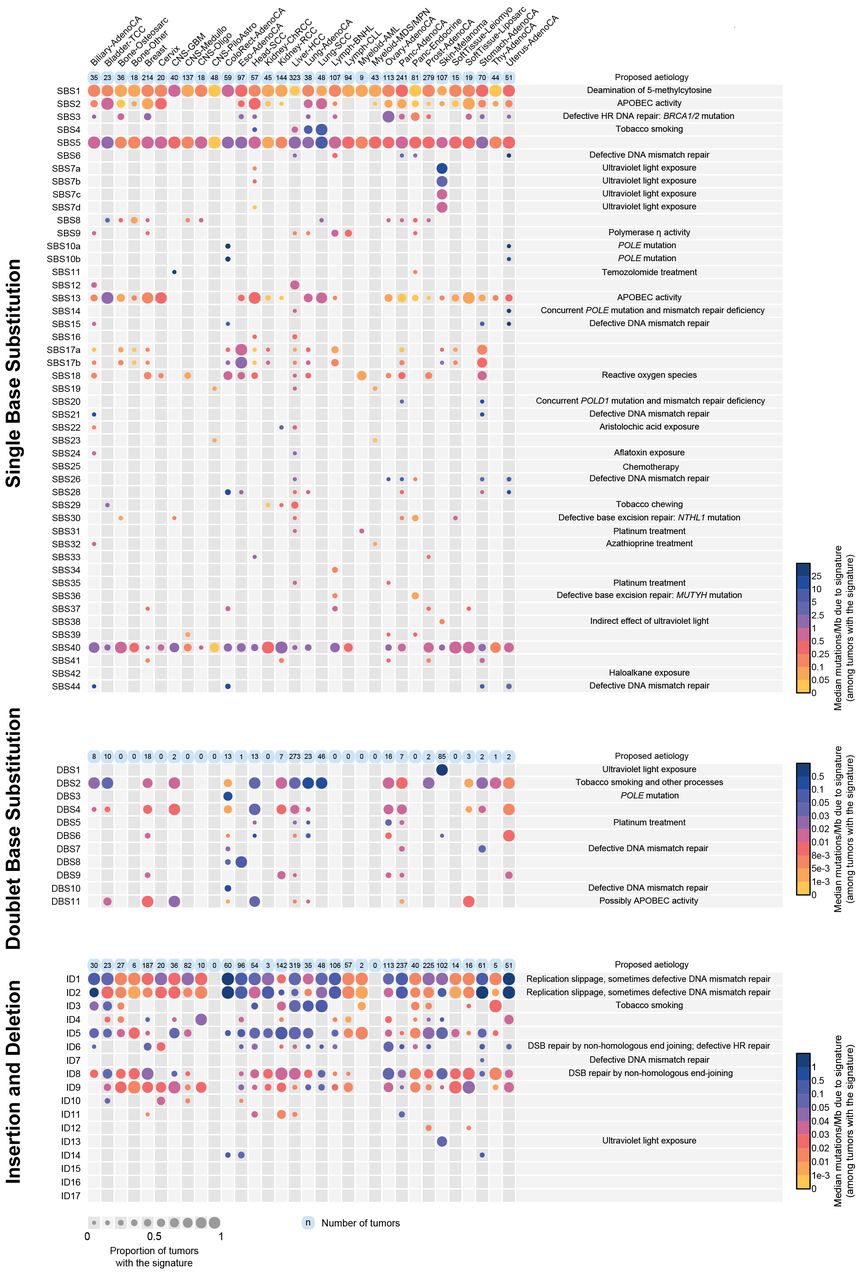
The number of mutations contributed by each mutational signature to the 2,780 PCAWG tumors. The numbers of mutations attributed are shown by cancer type. The size of each dot represents the proportion of samples of each tumor type that show the mutational signature. The colour of each dot represents the median mutation burden of the signature in samples which show the signature. Contributions are shown for single base substitution, doublet base substitution and indel mutational signatures separately. Contributions of composite signatures to the PCAWG cancers and single base substitution signatures to the complete set of cancer samples analysed are shown in Supplementary information.
Three previously characterised base substitution signatures (SBS7, SBS10, SBS17) split into multiple constituent signatures (Figure 2). We previously regarded SBS7 as a single signature composed predominantly of C>T at CCN and TCN trinucleotides (the mutated base is underlined) together with many fewer T>N mutations. It was found in malignant melanomas and squamous skin carcinomas and is likely due to UV induced pyrimidine dimer formation followed by translesion DNA synthesis by error-prone polymerases which predominantly insert adenine opposite damaged bases. With the larger dataset now available, SBS7 has decomposed into four constituent signatures: SBS7a consisting mainly of C>T at TCN; SBS7b consisting of C>T mainly at CCN and to a lesser extent at TCN; SBS7c and SBS7d, which constituted relatively minor components of the previous SBS7 and consist predominantly of T>A at NTT and T>C at NTT respectively42. Splitting of a mutational signature likely reflects the existence of multiple distinct mutational processes, initiated by the same exposure, which have closely, but not perfectly, correlated activities. For example, the constituent signatures of SBS7 are probably all initiated by UV-induced DNA damage. SBS7a and SBS7b may reflect different dipyrimidine photoproducts whereas SBS7c and SBS7d may be due to low frequencies of misincorporation by translesion polymerases of T and G opposite thymines in pyrimidine dimers rather than the more frequent and non-mutagenic A. Splitting of SBS10 and SBS17 is described at https://www.synapse.org/#!Synapse:syn12009783.
Several base substitution signatures showed transcriptional strand bias (https://www.synapse.org/#!Synapse:syn12009767). Transcriptional strand bias is often attributable to transcription coupled nucleotide excision repair (TC-NER) acting on DNA damaged by exogenous exposures which cause covalently bound bulky adducts or crosslinking to other bases and consequent distortion of the helical structure. This results in stalling of RNA polymerase and hence recruitment of the TC-NER machinery. An excess of DNA damage on untranscribed compared to transcribed strands of genes may also contribute to transcriptional strand bias43. Both mechanisms, however, result in more mutations of a damaged base on the untranscribed compared to the transcribed strands of genes. Assuming that either or both are responsible for the observed transcriptional strand biases (which may not always be the case), DNA damage to cytosine (SBS7a, SBS7b), guanine (SBS4, SBS8, SBS19, SBS23, SBS24, SBS31, SBS32, SBS35, SBS42), thymine (SBS7c, SBS7d, SBS21, SBS26, SBS33) and adenine (SBS5, SBS12, SBS16, SBS22, SBS25) may underlie these mutational signatures (see https://www.synapse.org/#!Synapse:syn12009783 for plots of strand bias). Although the likely underlying DNA damaging agents are known for SBS4 (tobacco mutagens), SBS7a, SBS7b, SBS7c, SBS7d (UV), SBS22 (aristolochic acid), SBS24 (aflatoxin), SBS25 (prior chemotherapy), SBS31 and SBS35 (platinum compounds), SBS32 (azathioprine), and SBS42 (haloalkanes), the causes of the remainder are unknown. Indeed, some signatures showing transcriptional strand bias are associated with defective DNA mismatch repair (SBS21 and SBS26) and it is conceivable that, for these, exogenous DNA damage is not involved. The extent of transcriptional strand bias appears to differ in different sectors of the genome. For example, consideration of the whole transcribed genome showed absent or minimal transcriptional strand bias in the APOBEC related SBS2 and SBS13 and in the defective polymerase epsilon proof-reading related SBS10a. However, consideration of exons alone showed clear evidence of transcriptional strand bias in these signatures (https://www.synapse.org/#!Synapse:syn12009783). The mechanism(s) underlying this amplification of transcriptional strand bias in exons is unknown and appears to be signature specific, since there is minimal difference in the extent of transcriptional strand bias between exons and other transcribed regions for other signatures (for example, SBS4).
Employing the single base substitution classification of 1536 mutation types, which uses the pentanucleotide sequence context two bases 5’ and two bases 3’ to each mutated base, yielded a set of signatures largely consistent with that based on substitutions in trinucleotide context alone. Notably, however, the pentanucleotide context enabled the extraction of two forms of both SBS2 and SBS13, one with mainly a pyrimidine (C or T) and the other with a purine (A or G) at the −2 base (the second base 5’ to the mutated cytosine). These may represent the activities of the cytidine deaminases APOBEC3A and APOBEC3B, respectively44. If so, APOBEC3A accounts for many more mutations than APOBEC3B in cancers with high APOBEC activity. Several other signatures showed non-random sequence contexts at +2 and −2 positions. In particular, the −2 bases in SBS17a and SBS17b and the −2 and +2 bases in SBS9 were predominantly A and T. In general, however, sequence context effects were much stronger for bases immediately 5′ and 3′ to the mutated bases.
SBS signatures showed substantial variation in the numbers of cancer types and cancer samples in which they were found, ranging from SBS1 and SBS5 which were present in almost every cancer type and almost every cancer sample, to SBS23 which was only observed in a small subset of liver cancers (Figure 3). The numbers of mutations per cancer sample attributed to each signature also varied greatly, from a few tens of mutations for SBS1 to millions of mutations for SBS10b. Almost all individual cancer samples exhibited multiple signatures, with a mode of three signatures per sample in the PCAWG set (https://www.synapse.org/#!Synapse:syn12169204). The assigned signatures reconstruct well the mutational spectra of the cancer samples (in PCAWG samples, median cosine similarity 0.97; 96.3% of samples with cosine similarity >0.90) (illustrative examples are shown in Figure 4).

Illustrative examples of mutational spectra of individual cancer samples (a breast cancer, a lung cancer and a malignant melanoma) and their contributory single base substitution, doublet base substitution and small insertion and deletion mutational signatures. Box initially at rest on sled sliding across ice.
Clustered single base substitution mutational signatures
Some mutational processes generate mutations that cluster in small regions of the genome. The relatively limited number of mutations generated by such processes, compared to those acting genome-wide, may result in failure to detect their signatures by standard methods. To obviate this problem, we first identified clustered mutations in each genome and analysed these separately (Methods). Four main signatures associated with clustered mutations were identified (Figure 2). Two found in multiple cancer types were similar to single base substitution SBS2 and SBS13, which have been attributed to APOBEC enzyme activity (mostly APOBEC3B) and represent foci of kataegis17,45 (Methods). Two additional clustered mutational signatures, one characterised by C>T and C>G mutations at (A|G)C(C|T) trinucleotides46 and the other T>A and T>C mutations at (A|T)T(A|T) were found in lymphoid neoplasms and likely represent direct and indirect consequences of activation induced cytidine deaminase (AID) mutagenesis (SBS84 and SBS85 respectively)6. The possibility that further processes may generate clustered mutations is not excluded.
Doublet base substitution (DBS) mutational signatures
Tandem doublet, triplet, quadruplet, quintuplet, and sextuplet base substitutions (https://www.synapse.org/#!Synapse:syn11801938, https://www.synapse.org/#!Synapse:syn11726620) at immediately adjacent bases were observed at ~1% the prevalence of single base substitutions. In most cancer genomes, the observed number of DBSs was considerably higher than expected from random adjacency of SBSs (https://www.synapse.org/#!Synapse:syn12177057) indicating the existence of commonly occurring, single mutagenic events that cause substitutions at neighbouring bases. There was substantial variation in the number of DBSs, ranging from zero to 20,818 in a sample. Across cancer types, the numbers of DBSs were generally proportional to the numbers of SBSs in that cancer type (Figure 1). However, colorectal adenocarcinomas had significantly fewer DBS than expected, and lung cancers and melanomas had more (Extended Data Table 1). The large dataset analysed here allowed, for the first time, systematic analysis of DBS and indel signatures (described below). Eleven DBS signatures were extracted (Figure 2).
DBS1 was characterised almost exclusively by CC>TT mutations (Figure 2), contributed 100s-10,000s of mutations in malignant melanomas (Figure 3) with SBS7a and SBS7b. DBS1 exhibited transcriptional strand bias consistent with damage to cytosines (https://www.synapse.org/#!Synapse:syn12177063). CC>TT mutations associated with UV induced DNA damage are well established in the literature and are thought to be due to generation of pyrimidine dimers and subsequent error-prone translesion DNA synthesis by polymerases that introduce adenines opposite the damaged bases47.
Reanalysis after exclusion of malignant melanomas and other cancers with evidence of UV exposure still yielded a signature (termed DBS11) characterised predominantly by CC>TT mutations and smaller numbers of other doublet base substitutions at CC and TC which contributed 10s of mutations to many samples of multiple cancer types (Figures 2 and 3). DBS11 was associated with SBS2 which is due to APOBEC activity. Thus, APOBEC activity may also generate DBS11, although the mechanism by which it induces doublet base substitutions is not well understood.
DBS2 was composed predominantly of CC>AA mutations, with smaller numbers of CC>AG and CC>AT mutations, and contributed 100s-1000s of mutations in lung adenocarcinoma, lung squamous and head and neck squamous carcinomas, which are often caused by tobacco smoking (Figures 2 and 3). DBS2 showed transcriptional strand bias indicative of guanine damage (https://www.synapse.org/#!Synapse:syn12177064) and was associated with SBS4 which is caused by tobacco smoke exposure. It is likely, therefore, that DBS2 can be a consequence of DNA damage by tobacco smoke mutagens.
Analysis of each cancer type separately, however, revealed a signature very similar to DBS2 contributing 100s of mutations to liver cancers and 10s of mutations to cancers of other types without evidence of tobacco smoke exposure. A pattern closely resembling DBS2 and characterised predominantly by CC>AA mutations, together with smaller contributions of CC>AG and CC>AT, dominates DBSs in normal mouse cells and is particularly frequent in the liver48. The nature of the mutational processes underlying these doublet signatures in smoking-unrelated human cancers and in normal mice is unknown. However, acetaldehyde exposure in experimental systems generates a mutational signature characterised primarily by CC>AA and lower burdens of CC>AG and CC>AT mutations together with single base substitution C>A mutations49. Acetaldehyde is an oxidation product of alcohol and a constituent of cigarette smoke. The role of acetaldehyde, and perhaps other aldehydes, in generating DBS2, whether associated with tobacco smoking, alcohol consumption or in nonexposed cells, merits further investigation50.
DBS3, DBS7, DBS8 and DBS10 showed 100s-1000s of mutations in rare colorectal, stomach and oesophageal cancers some of which showed evidence of defective DNA mismatch repair (DBS7, DBS10) or polymerase epsilon exonuclease domain mutations (DBS3) generating hypermutator phenotypes (Figures 2, 3). DBS5 was found in cancers previously exposed to platinum chemotherapy and is associated with SBS31 and SBS35. The remaining DBS signatures are of uncertain cause.
Small insertion and deletion (ID) mutational signatures
Indels were usually present at ~10% the frequency of base substitutions (Figure 1). There was substantial variation between cancer genomes in numbers of indels, even when cancers with evidence of defective DNA mismatch repair were excluded. Overall, the numbers of deletions and insertions were similar, but there was variation between cancer types with some showing more deletions and others more insertions of various subtypes (Figure 1). Seventeen indel mutational signatures were extracted (Figure 2).
Indel signature 1 (ID1) was composed predominantly of insertions of thymine and ID2 of deletions of thymine, both at long (≥5) thymine mononucleotide repeats (Figure 2). 10s to 100s of mutations of both signatures were found in the large majority of most cancer types but were particularly common in colorectal, stomach, endometrial and oesophageal cancers and in diffuse large B cell lymphoma (Figure 3). Most of these cancers are likely to be DNA mismatch repair proficient on the basis of the relatively limited numbers of indels and absence of the SBS signatures (SBS6, SBS14, SBS15, SBS20, SBS21, SBS26, and SBS44) associated with DNA mismatch repair deficiency. Together, ID1 and ID2 accounted for 97% and 45% of indels in hypermutated and non-hypermutated cancer genomes, respectively (Extended Data Table 2), and both signatures have also been found in non-neoplastic cells51. They are likely due to the intrinsic tendency to slippage during DNA replication of long mononucleotide tracts. However, the mechanistic basis for separation into two signatures, one presumably due to slippage of the nascent strand (ID1) and the other the template strand (ID2) is unclear. Similarly, the substantial differences in their mutation frequencies between cancer types are not well understood.
ID3 was characterised predominantly by deletions of cytosine at short (≤5bp long) mononucleotide cytosine repeats and exhibited 100s of mutations in tobacco smoking associated cancers of the lung and head and neck (Figures 2 and 3). There was transcriptional strand bias of mutations, with more guanine deletions than cytosine deletions on the untranscribed strands of genes, compatible with TC-NER of adducted guanine (https://www.synapse.org/#!Synapse:syn12177065, https://www.synapse.org/#!Synapse:syn12177066). The numbers of ID3 mutations in cancer samples positively correlated with the numbers of SBS4 and DBS2 mutations, both of which have been associated with tobacco smoking (Extended Data Figure 6). It is therefore likely that DNA damage by components of tobacco smoke underlie ID3 but the mechanism(s) by which indels are generated is unclear.
ID13 was characterised predominantly by deletions of thymine at thymine-thymine dinucleotides and exhibited large numbers of mutations in malignant melanomas of the skin (Figures 2 and 3). The numbers of ID13 mutations correlated with the numbers of SBS7a, SBS7b and DBS1 mutations, which have been attributed to DNA damage induced by UV (Extended Data Figure 6). It is, however, notable that a similar mutation of the other pyrimidine, ie deletion of cytosine at cytosine-cytosine dinucleotides, does not feature strongly in ID13, perhaps reflecting the predominance of thymine compared to cytosine dimers induced by UV52. The mechanism(s) underlying thymine deletion is unclear.
ID6 and ID8 were both characterised predominantly by deletions ≥5bp (Figure 2). ID6 exhibited overlapping microhomology at deletion boundaries with a mode of 2bp and often longer stretches. This signature was correlated with SBS3 which has been attributed to defective homologous recombination based repair (Extended Data Figure 6). By contrast, ID8 deletions showed shorter or no microhomology at deletion boundaries, with a mode of 1bp, and did not strongly correlate with SBS3 mutations (Figures 2 and 3). These indel patterns are characteristic of DNA double strand break repair by non-homologous recombination based end-joining mechanisms and indicate that at least two distinct forms of end-joining mechanism are operative in human cancer53.
A small fraction of cancers exhibited very large numbers of ID1 and ID2 mutations (>10,000) (Figure 3, https://www.synapse.org/#!Synapse:syn12009775). These were usually accompanied by SBS6, SBS14, SBS15, SBS20, SBS21, SBS26 and/or SBS44 which are associated with DNA mismatch repair deficiency, sometimes combined with POLE or POLD1 proofreading deficiency (SBS14, SBS20)36. Occasional cases with these signatures additionally showed large numbers of ID7 indels (https://www.synapse.org/#!Synapse:syn11738668). In addition, rare samples showed large numbers of either ID4, ID11, ID14, ID15, ID16 or ID17 mutations but did not show ID1 and ID2 mutations or the single base substitution signatures usually associated with DNA mismatch repair deficiency. The mechanisms underlying these signatures are unknown.
Composite mutational signatures
In the analyses described above mutational signatures were extracted for each mutation type separately. However, mutational processes in nature generate composite signatures that may include SBSs, DBSs, IDs, genome rearrangements and chromosome number changes. We therefore also extracted signatures using combined catalogues of SBSs, DBSs, and IDs (257 mutation subclasses or 1697 if the 1536 classification of single base substitutions was used). Fifty two composite signatures were extracted.
A composite signature with components similar to SBS4, DBS2 (characterised predominantly by CC>AA mutations) and ID3 (characterised predominantly by deletion of cytosine at short runs of cytosines) was found mainly in lung cancers, suggesting that it is the consequence of tobacco smoke exposure (Extended Data Figure 7). Similarly, composite signatures with components similar to SBS7a, SBS7b, DBS1 (characterised predominantly by CC>TT mutations) and ID13 (characterised predominantly by deletion of thymine at thymine–thymine dinucleotides) were found in skin cancers and are thus likely due to UV induced DNA damage (Extended Data Figure 7). A further composite signature in breast and ovarian cancers included features of SBS3 and ID6 combined with ID8 (deletions >5bp with varying degrees of overlapping microhomology) and is likely associated with defective homologous recombination based repair (Extended Data Figure 7). In these composite signatures attributions of the constituent SBS, DBS and ID signatures extracted independently in the main analyses were correlated with each other, adding support to the existence of the composite signatures (Extended Data Figure 6). Various forms of defective DNA mismatch repair were also associated with multiple SBS, DBS and ID signatures.
Correlations with age
A positive correlation between age of cancer diagnosis and the number of mutations attributable to a signature suggests that the mutational process underlying the signature has been operative, at a more or less constant rate, throughout the cell lineage from fertilized egg to cancer cell, and thus in normal cells from which that cancer type develops4,54. Confirming previous reports, the numbers of SBS1 and SBS5 mutations correlated with age, exhibiting different rates in different tissue types (Q values in https://www.synapse.org/#!Synapse:syn12030687, https://www.synapse.org/#!Synapse:syn12217988). In addition, SBS40 correlated with age in multiple cancer types. However, given the similarity in signature profile between SBS5 and SBS40 the possibility of misattribution between these signatures cannot currently be excluded. The numbers of DBSs and IDs were much lower than the numbers of SBSs and the numbers of samples in which DBS and ID signatures could be attributed were also lower. Nevertheless, DBS2 and DBS4 correlated with age and, consistent with the interpretation of activity in normal cells, the profiles of DBS2 and DBS4 together closely resemble the spectrum of DBS mutations found in normal mouse cells48. Neither DBS2 nor DBS4, however, was clearly correlated with an SBS or ID signature that correlates with age. ID1, ID2, ID5 and ID8 showed correlations with age in multiple tissues. ID1 and ID2 indels are likely due to slippage at poly T repeats during DNA replication and correlated with the number of SBS1 substitutions. SBS1 has previously been proposed to reflect the number of mitoses a cell has experienced and thus SBS1, ID1 and ID2 may all be generated during DNA replication at mitosis4. The number of ID5 mutations correlated with the number of SBS40 mutations and thus the mutational processes underlying these two age correlated signatures may also harbour common components. ID8 is predominantly composed of deletions >5bp with no or 1bp of microhomology at their boundaries. These are likely due to DNA double strand breaks which have not been repaired by homologous recombination based mechanisms, but instead by a non-homologous-end joining mechanism. The features of ID8 resemble those of some ionising radiation associated mutations and this may, therefore, be an underlying aetiological factor55. Taken together, the results indicate that multiple mutational processes operate in normal cells.
DISCUSSION
Cancers arise as a result of somatic mutations. Mutational signature analysis therefore provides important insights into cancer development through comprehensive characterisation of the underlying mutational processes. There are, however, important constraints, limitations and assumptions in the analytic frameworks we have used that should be recognised. Although designed to reflect the mutational consequences of recurrent mutational processes, mutational signatures extracted from sample sets in which multiple mutational processes are operative remain mathematical approximations, with profiles that can be influenced by the mathematical approach used and by additional factors, such as the other mutational processes present. For conceptual and practical simplicity we have assumed that there is a single signature associated with each mutational process and have provided an average, reference signature to represent it. However, we do not discount the possibility that further nuances and variations of signature profiles exist, for example between different tissues. Moreover, although the extent of separation between partially correlated signatures has been improved in this analysis, some signatures may still represent combinations of constituent signatures. Contributions from each signature to the burden of mutations in each sample have been estimated. However, with increasing numbers of signatures and multiple orders of magnitude differences in mutation burdens from certain signatures, prior knowledge can help to avoid biologically implausible results. Thus further development of methods for deciphering mutational signatures and attribution of mutations is warranted and this needs to be supplemented by signatures derived from experimental systems in which the causes of the mutations are known. The numbers of DBSs, clustered substitutions, IDs and genome rearrangements (reported in 30) are small compared to single base substitutions. Thus, larger datasets may be required to robustly characterise their mutational signatures. Nevertheless, the results outlined here indicate that signatures with many similarities and some differences can be found by different mathematical approaches, and that these are confirmed in many different ways, including experimentally elucidated signatures22,31,38,41,42,54,56–62 and the observation of tumors dominated by a single signature (https://www.synapse.org/#!Synapse:syn12016215)
This analysis includes almost all publicly available exome and whole-genome cancer sequences, amounting in aggregate to 23,829 cancers of most cancer types. Some rare or geographically restricted signatures may not have been captured and signatures of therapeutic mutagenic exposures have not been exhaustively explored. Nevertheless, it is likely that a substantial proportion of the naturally-occurring mutational signatures found in human cancer have now been described. This comprehensive repertoire provides a foundation for future research into (i) geographical and temporal differences in cancer incidence to elucidate underlying differences in aetiology, (ii) the mutational processes and signatures present in normal tissues and caused by non-neoplastic disease states, (iii) clinical and public health applications of signatures as indicators of sensitivity to therapeutics and past exposure to mutagens, and (iv) mechanistic understanding of the mutational processes underlying carcinogenesis.
Acknowledgements
The results here are partly based on data generated by the TCGA Research Network (http://cancergenome.nih.gov/).
This work was supported by Wellcome grant reference 206194 (M.R.S., S.M., L.B.A.), <G.G. funding>, Singapore National Medical Research Council grant NMRC/CIRG/1422/2015 and the Singapore Ministry of Health via the Duke-NUS Signature Research Programmes (M.N.H., A.W.T.N., A.B., S.G.R.), US National Institute of Health Intramural Research Program Project Z1AES103266 (D.A.G.), the European Research Council Consolidator Grant 682398 (N.L.-B.), US National Cancer Institute U24CA143843 (D.A.W.), and. Cancer Research UK Grand Challenge Award C98/A24032 (L.B.A., M.R.S.). We thank XXX





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}